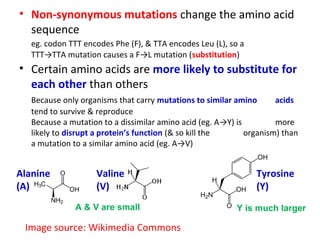
- The document discusses alignment scoring functions used to score alignments of protein sequences.
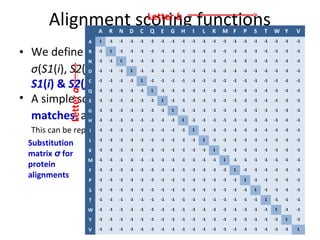
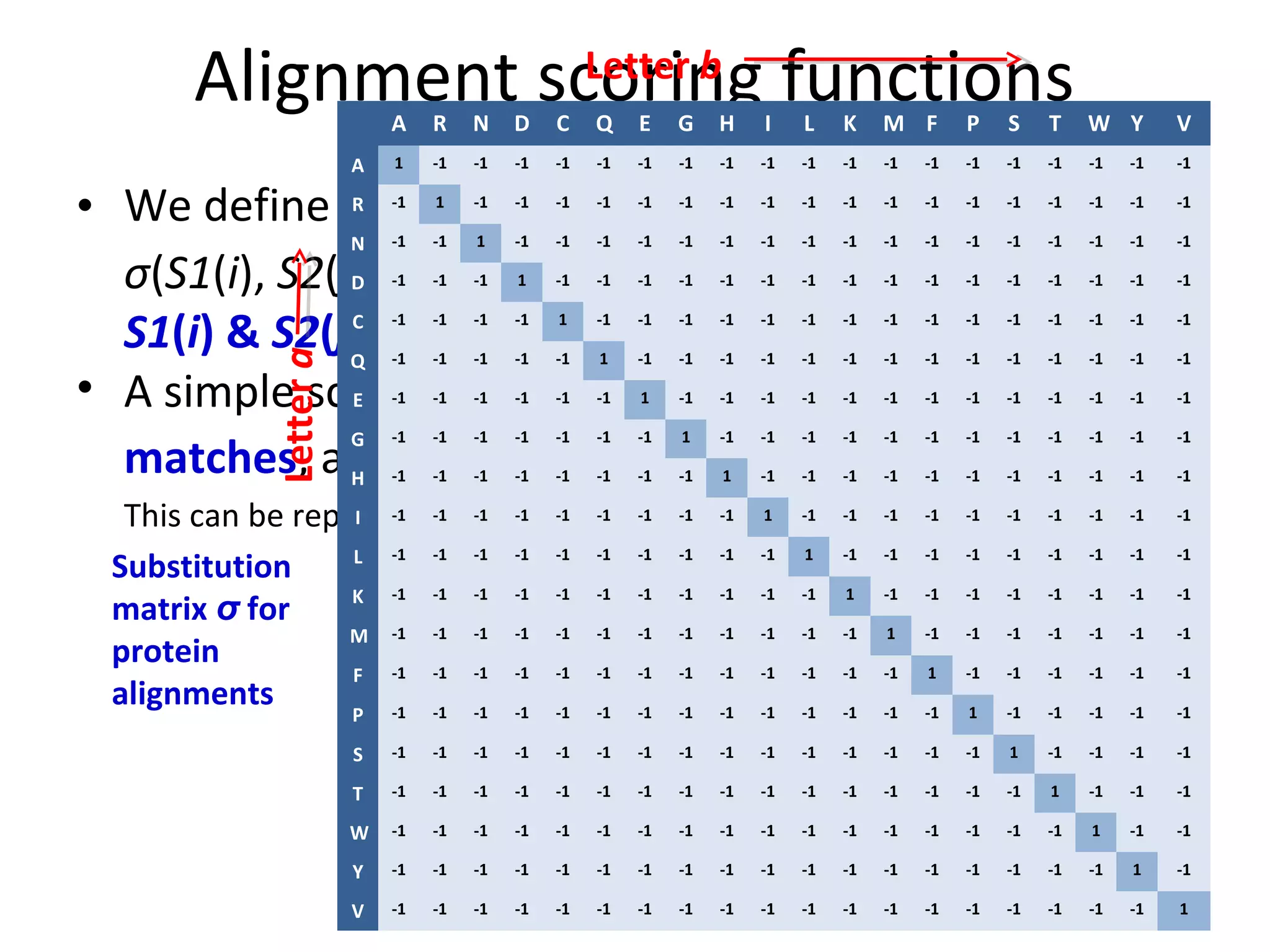
- A simple scoring function gives a score of +1 for matches and -1 for mismatches. This can be represented as a substitution matrix.
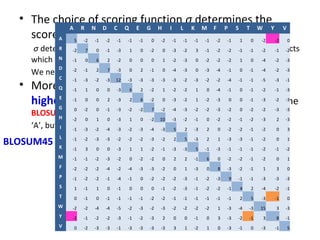
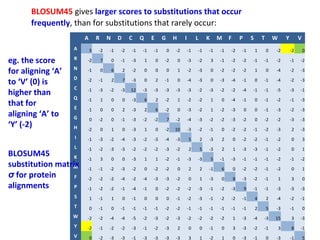
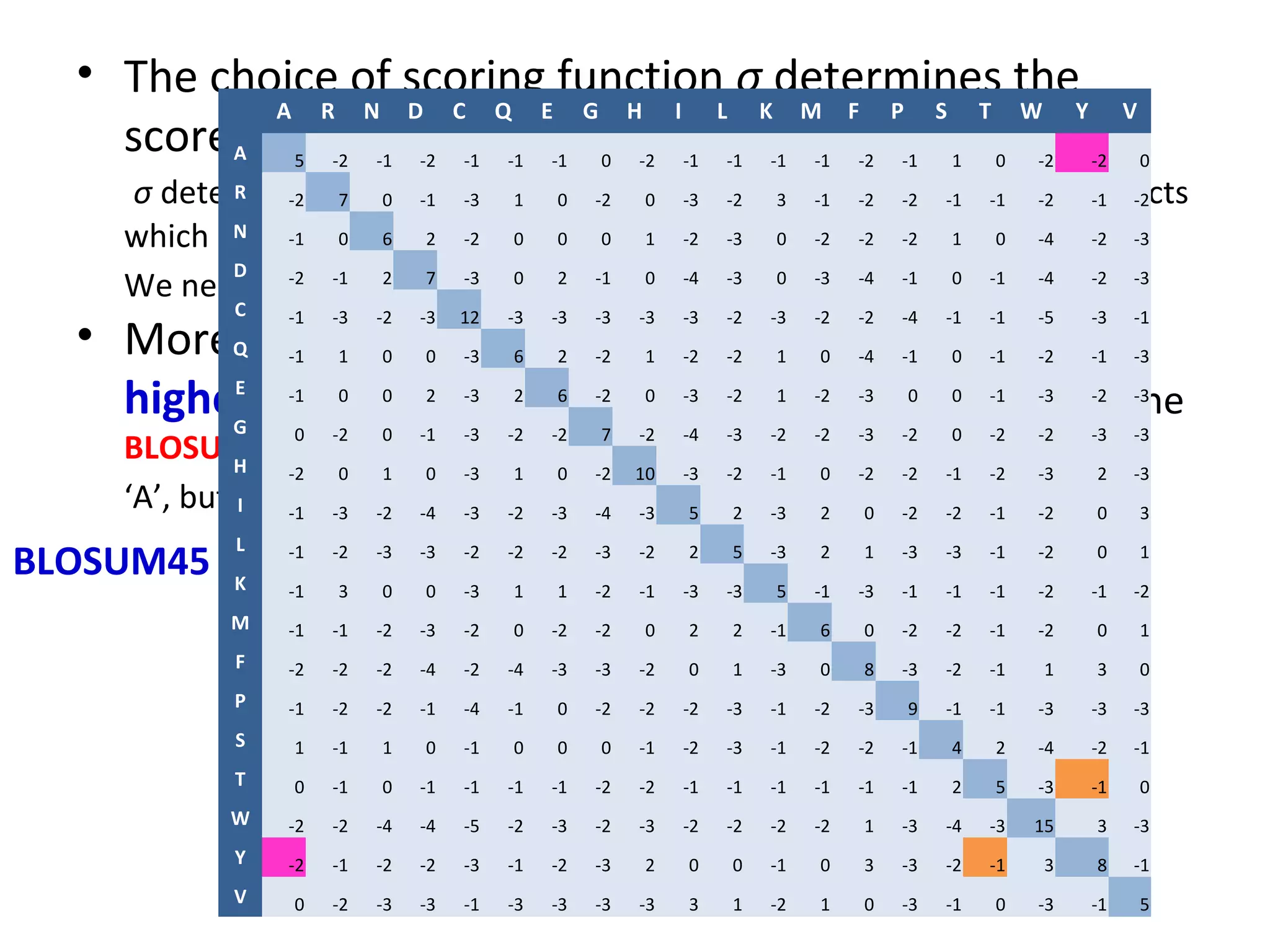
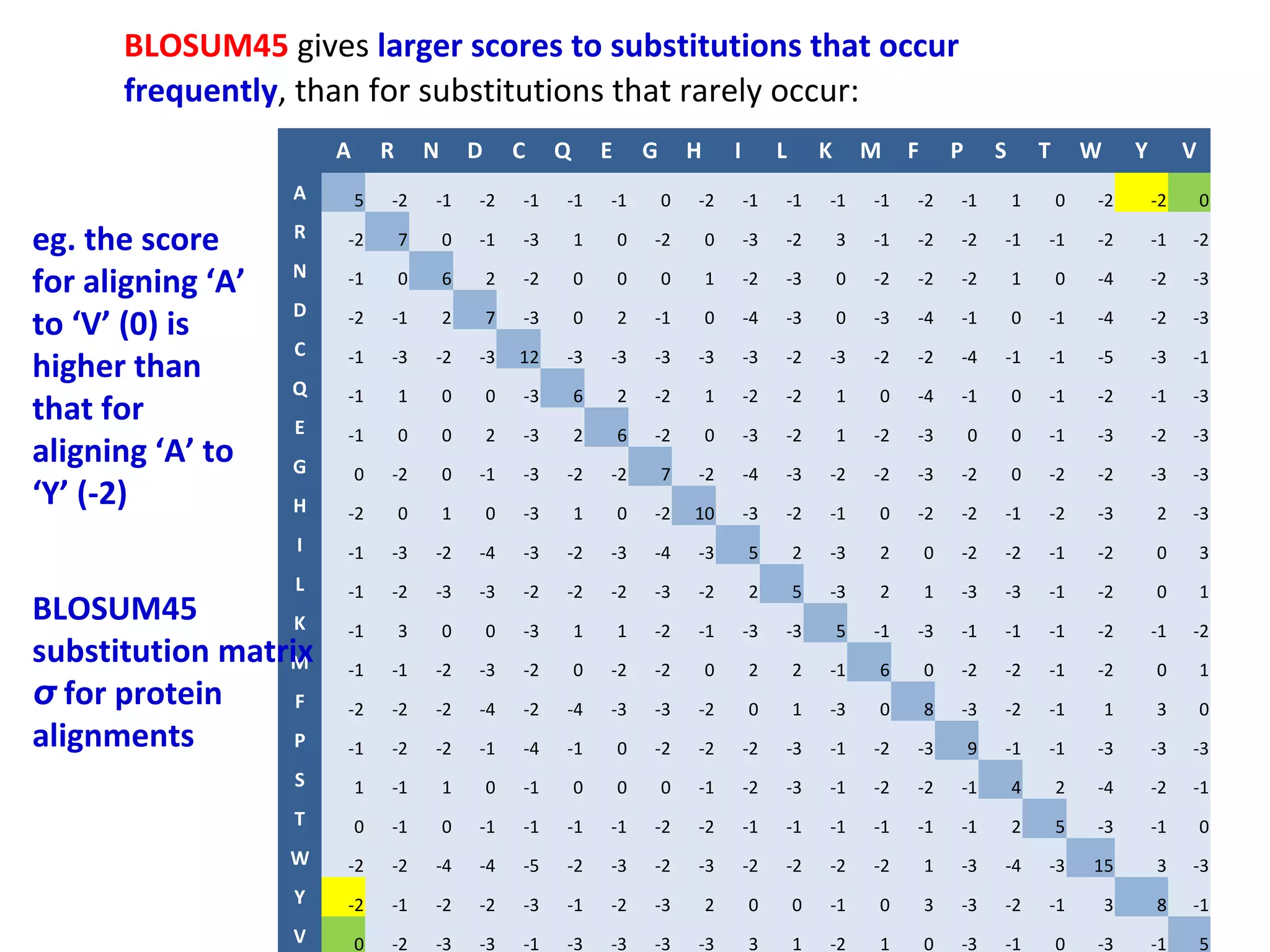
- More complex scoring functions like BLOSUM45 take into account that certain amino acid substitutions are more likely than others based on evolution.
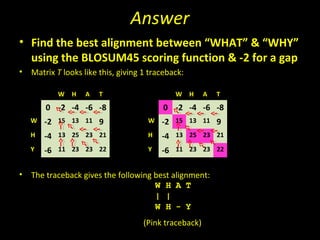
- The choice of scoring function determines which alignments are considered "best" or highest scoring.