Downloaded 210 times





































This document provides an overview of bioinformatics and some of its key applications. It discusses how bioinformatics is an interdisciplinary field that uses computer science, statistics and other approaches to analyze large amounts of biological data. It notes that bioinformatics has become necessary due to the explosion of genomic data from projects like the Human Genome Project. Some of the goals and uses of bioinformatics mentioned include uncovering biological information from data, applications in molecular medicine, agriculture and environmental science. The document also provides brief descriptions of structural bioinformatics, common biological databases, MASCOT database searching, and scoring schemes used in bioinformatics.
Overview of bioinformatics, its search tools like MASCOT and UniProt, setting the stage for further discussion.
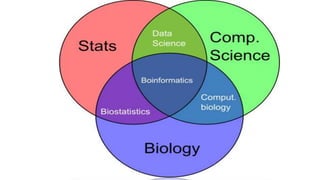
Definition and significance of bioinformatics. Highlights interdisciplinary nature and areas where it contributes significantly.
Focuses on structural bioinformatics and notable databases like GenBank and UniProt for research and data.
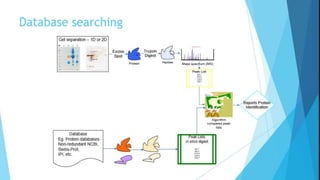
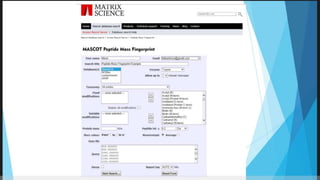
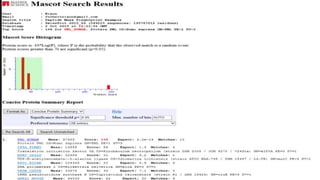
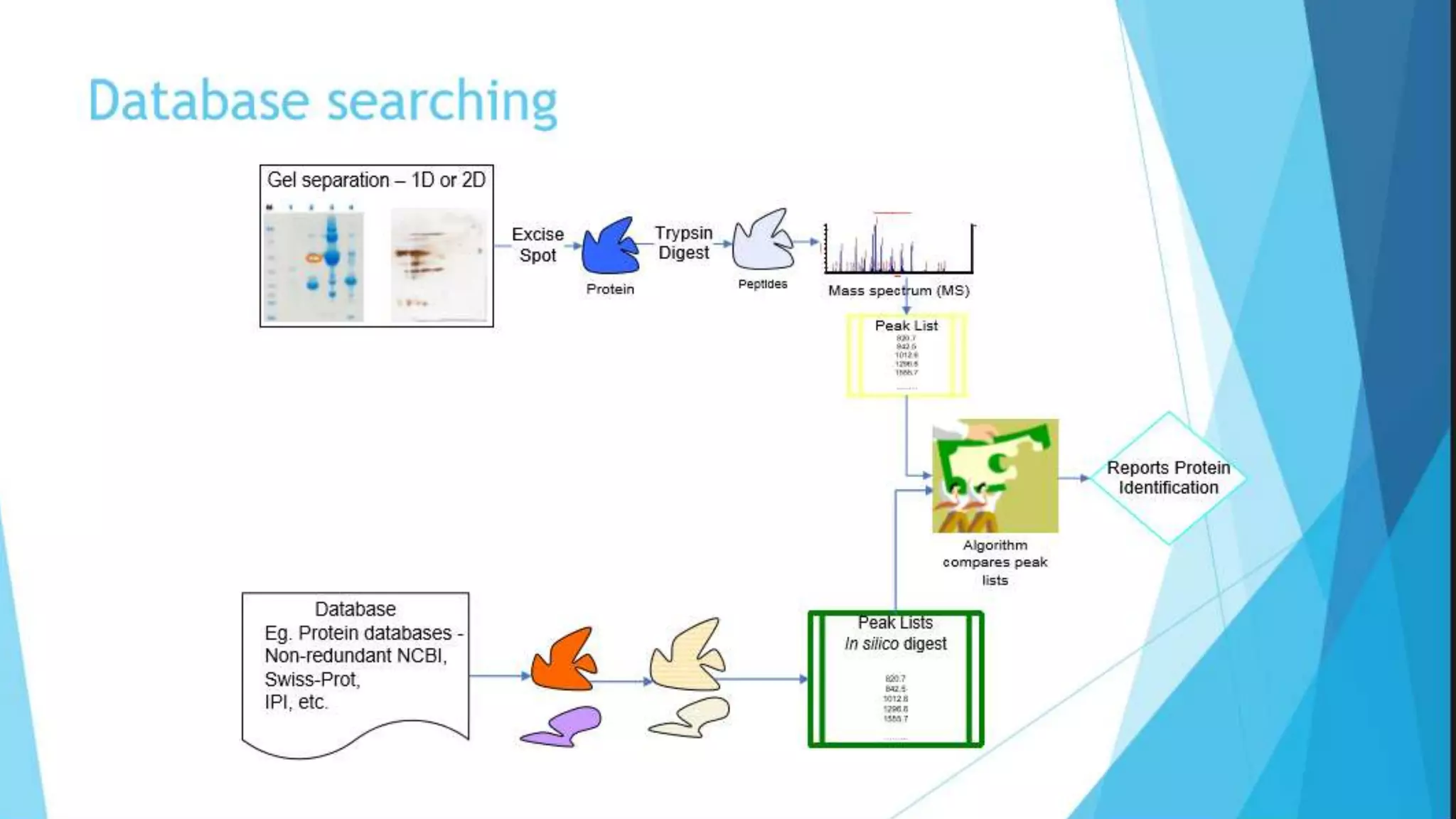
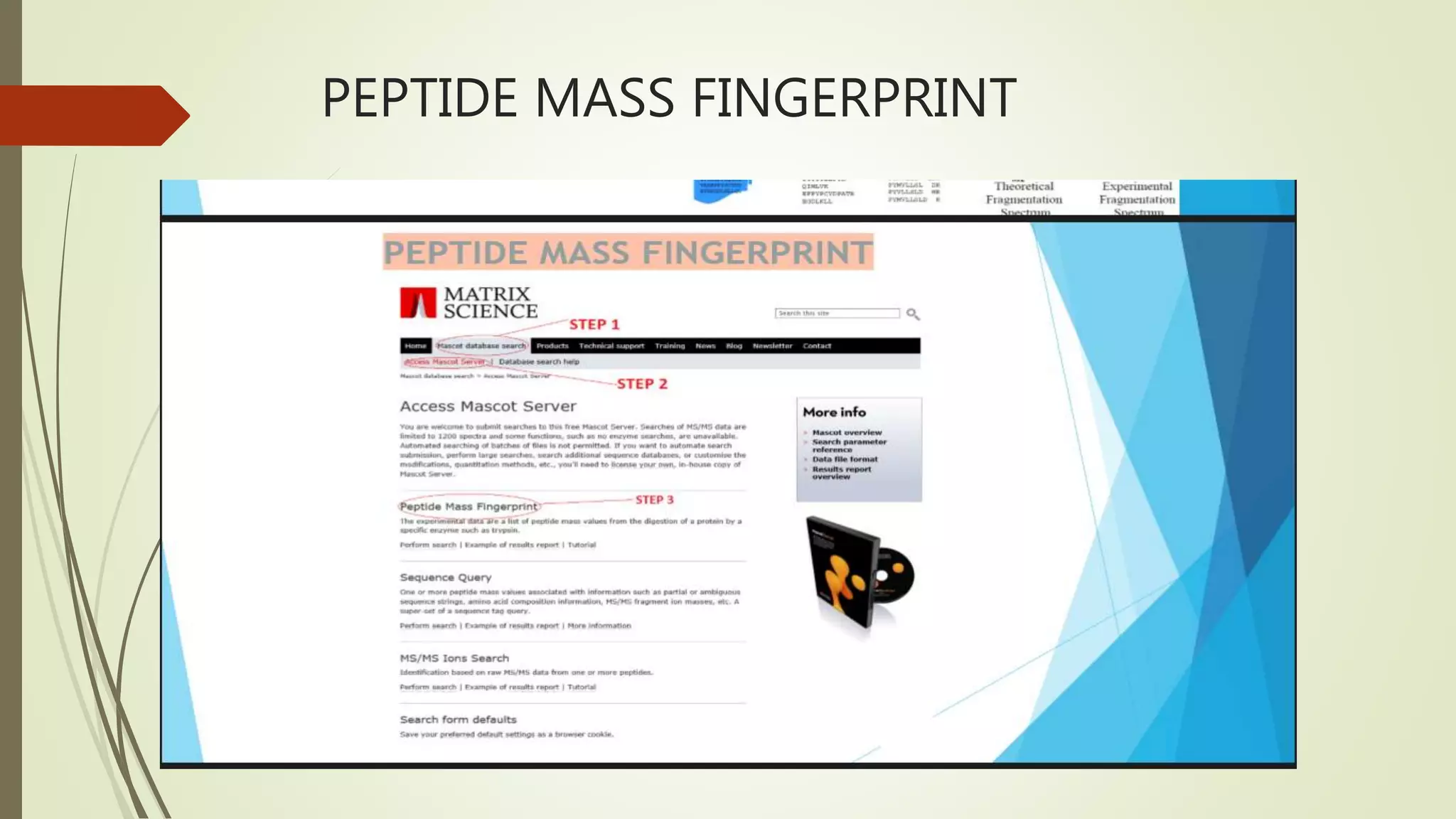
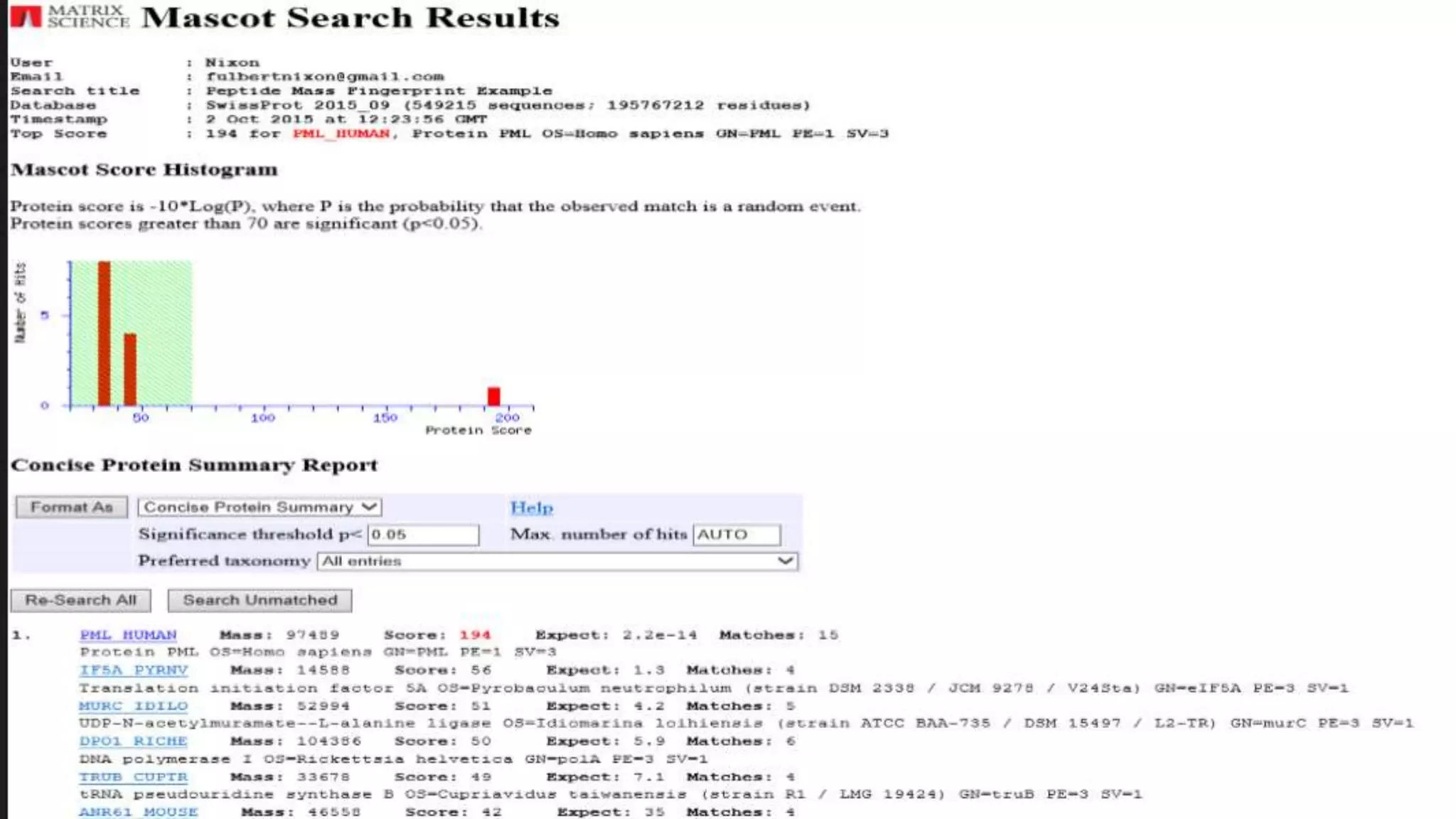
Explains MASCOT’s role in interpreting mass spectral data for protein identification and its capabilities.
Details the MOWSE algorithm used by MASCOT, scoring schemes, and parameters for effective database searching.
Final slide thanking the audience, closing the presentation.























































































