ZIAUDDIN UNIVERSITY
Faculty OfEngineering Science, Technology & Management
(ZUFESTM)
Department Of Biomedical Engineering
Dr. Syeda Bushra Zafar
Assistant Professor
Bioinformatics
Multiple Sequence Alignment
2.
Multiple sequence alignment
•Multiple sequence alignment is to align multiple related sequences to achieve
optimal matching of the sequences.
• There are unique advantage of multiple sequence alignment:
Reveals biological information
Carry out phylogenetic analysis
Designing of degenerate polymerase chain reaction primers
3.
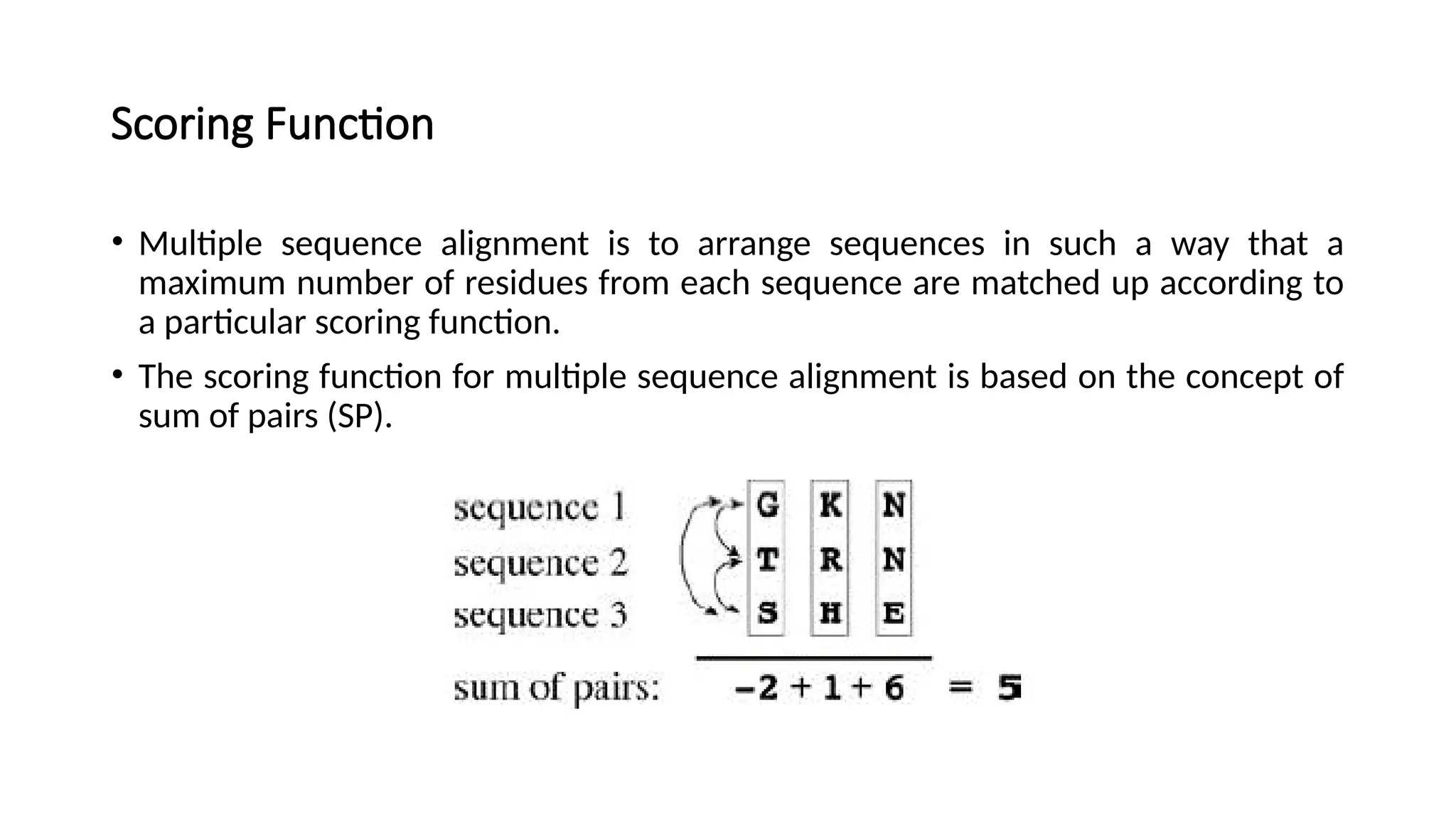
Scoring Function
• Multiplesequence alignment is to arrange sequences in such a way that a
maximum number of residues from each sequence are matched up according to
a particular scoring function.
• The scoring function for multiple sequence alignment is based on the concept of
sum of pairs (SP).
Exhaustive algorithm
• Theexhaustive alignment method involves examining all possible aligned positions simultaneously
• This approach is similar to dynamic programming, but dynamic programming is limited to small datasets of
less than ten short sequences.
• A program called, divide and conquer alignment (DCA) which uses some exhaustive components is used.
• Divide and conquer alignment:
• It is a semi exhaustive approach
• It works by breaking sequences into small fragments
• This algorithm provides an option of using a more heuristic procedure (fastDCA) to choose optimal cutting
points so it can more rapidly handle a greater number of sequences.
• The resulting short alignments are joined together head to tail
• When the length of sequences reach a predefined threshold then dynamic programming is applied for
alignment.
• It performs global alignment and requires the input sequences to be of similar lengths and domain structures.
6.
Heuristic algorithm
• Dynamicprogramming is not feasible for routine multiple sequence alignment, a
faster heuristic algorithm has been developed.
• Categories of heuristic algorithm:
1. Progressive alignment type
2. Iterative alignment type
3. Block-based alignment type
7.
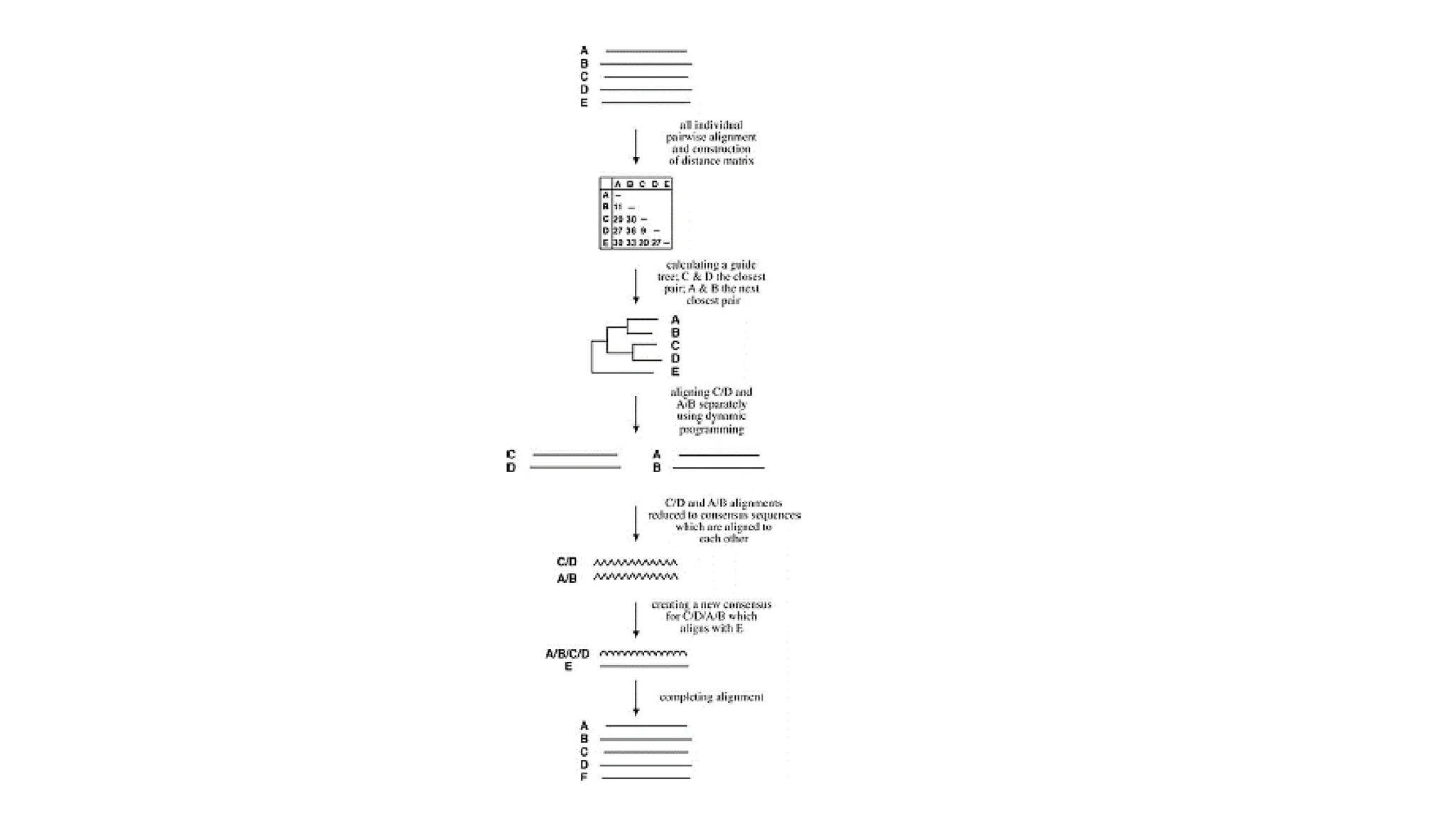
Progressive Alignment Method
•It first conducts pairwise alignments for each possible pair of sequences and generate
score
• The scores are then converted into evolutionary distances to generate a distance
matrix for all the sequences involved
• A phylogenetic tree is generated using the neighbor-joining method
• According to the guide tree, the two most closely related sequences are first re-aligned
• Two already aligned sequences are converted to a consensus sequence with gap
positions fixed. The consensus is then treated as a single sequence in the subsequent
step.
• The next closest sequence based on the guide tree is aligned with the consensus
sequence using dynamic programming.
9.
Clustal
• Probably themost well-known progressive alignment program is Clustal
• Clustal is a progressive multiple alignment program available either as a stand-
alone or on-line program.
• The stand-alone program, which runs on UNIX and Macintosh, has two variants,
Clustal W and Clustal X.
• One of the most important features of this program is the flexibility of using
substitution matrices.
• For closely related sequences BLOSUM62 or PAM120 matrix are used, whereas,
for more divergent sequences BLOSUM45 or PAM250 matrices may be used.
• Use of adjustable gap penalties that allow more insertions and deletions in
regions that are outside the conserved domains, but fewer in conserved regions
10.
• The programalso applies a weighting scheme to increase the reliability of aligning
divergent sequences
Drawbacks and Solutions
It is unsuitable for comparing sequences of different lengths. Accuracy is
compromised.
• Another major limitation is the “greedy” nature of the algorithm. The final
alignment could be far from optimal
11.
T-Coffee (Tree-based ConsistencyObjective Function for
alignment Evaluation)
• T-Coffee performs both global and local pairwise alignment for all possible pairs involved
The global pairwise alignment is performed using the Clustal program. The local pairwise
alignment is generated by the Lalign program, from which the top ten scored alignments
are selected
Both global and local alignments are pooled
Consistency of alignment is evaluated and a score is generated.
Each pairwise alignment is further aligned with a possible third sequence.
The result is used to refine the original pairwise alignment based on a consistency
criterion in a process known as library extension.
Distance matrix is built to derive a guide tree
Multiple sequence alignment performed using progressive approach.
12.
• T-Coffee avoidsthe problem of getting stuck in the suboptimal alignment regions,
because an optimal initial alignment is chosen from many alternative alignments
• T-Coffee indeed outperform Clustal however, it is slower than Clustal because of
the time required for calculation of consistency score.
• T-Coffee provides a graphical output of the alignment results.
13.
Iterative alignment
• Theiterative approach is based on the idea that an optimal solution can be found
by repeatedly modifying existing suboptimal solutions.
• The order of the sequences used for alignment is different in each iteration
• This method may alleviate the “greedy” problem of the progressive strategy,
because order is change everytime.
• This method does not have guarantees for finding the optimal alignment as it is
also heuristic in nature
14.
PRRN
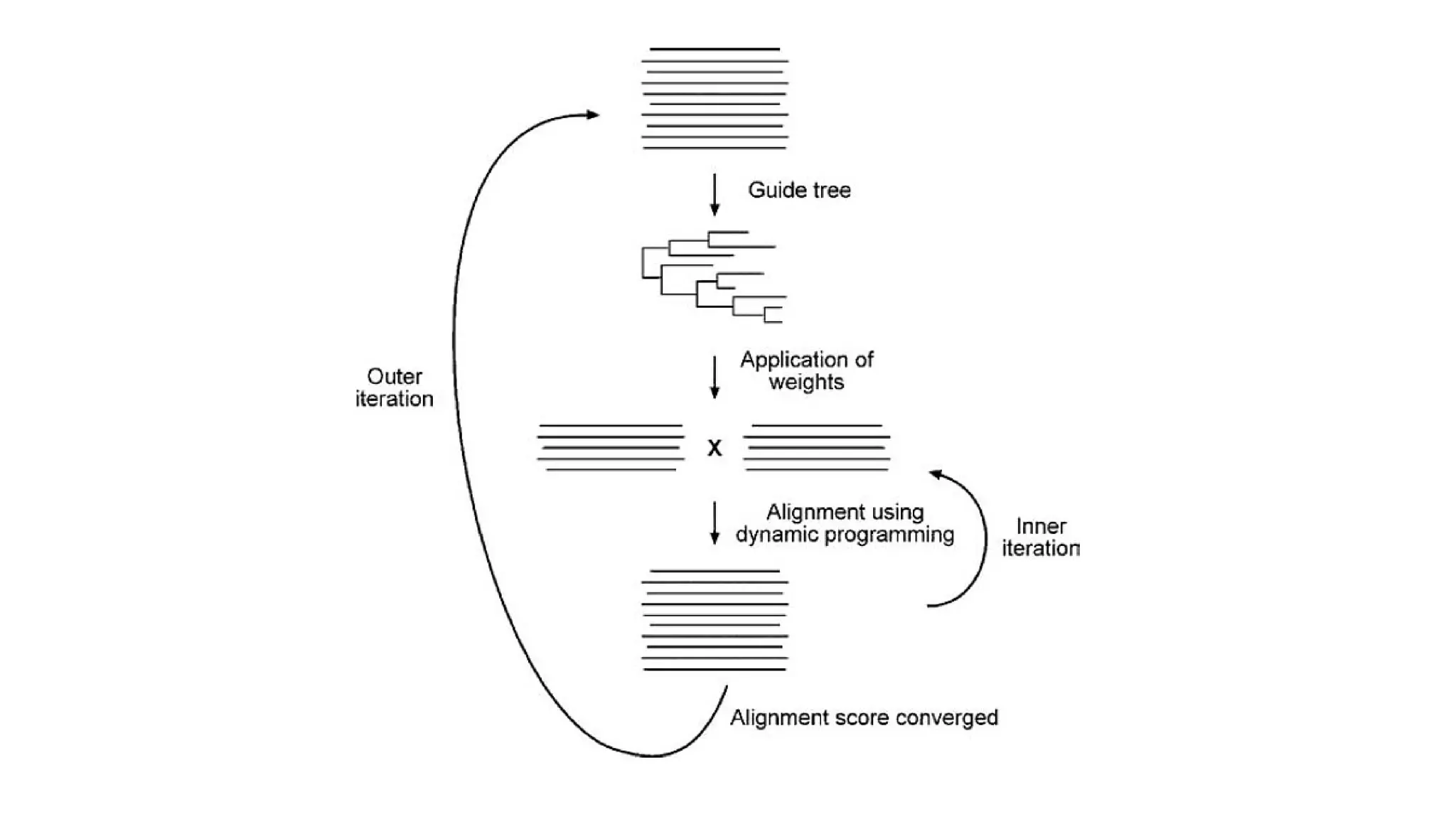
• Uses adouble nested iterative strategy for multiple alignment. It performs multiple alignment
through two sets of iterations: inner iteration and outer iteration
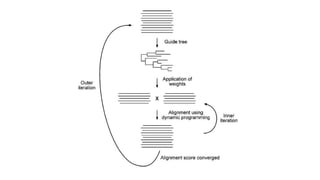
• In the outer iteration, an initial random alignment is generated that is used to derive a UPGMA tree
• In the inner iteration, first the sequences are divided randomly into two groups
• The two groups, each treated as a single sequence, are then aligned to each other using global
dynamic programming
• The process is repeated through many cycles until the total SP score no longer increases
• Tree is generated
• New weights are applied to optimize alignment scores. The newly optimized alignment is subject to
further realignment in the inner iteration.
• This process is repeated over many cycles until there is no further improvement in the overall
alignment scores
16.
Block-Based Alignment
• Itis a local alignment based strategy that identifies a block of ungapped
alignment shared by all the sequences.
DIALIGIN 2
• The method breaks each of the sequences down to smaller segments and
performs all possible pairwise alignments between the segments.
• High-scoring segments, called blocks, among different sequences are then
compiled in a progressive manner to assemble a full multiple alignment
• It places emphasis on block-to-block comparison rather than residue-to-residue
comparison
17.
Match-Box
• The programcompares segments of every nine residues of all possible pairwise
alignments.
• If the similarity of particular segments is above a certain threshold across all
sequences, they are used as an anchor to assemble multiple alignments