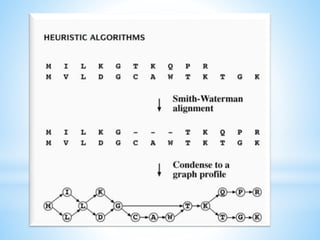
This document discusses different methods for multiple sequence alignment, including scoring functions, exhaustive and heuristic algorithms. It describes progressive alignment methods like Clustal that first conduct pairwise alignments and then progressively align more divergent sequences. Issues like getting stuck in suboptimal alignments are addressed by methods like T-Coffee. Block-based methods like DIALIGN2 and Match-Box identify conserved blocks shared across sequences. Manual editing may be needed to further improve automated alignments.
Multiple sequence alignment uses scoring functions and the sum of pairs method. BLOSUM62 matrix indicates alignment likelihood is 32 times more probable than random.
Alignment methods include exhaustive and heuristic algorithms. Clustal is a notable progressive alignment program, using different scoring matrices based on sequence similarity.
Block-based alignment methods detect conserved domains. Programs like DIALIGN2 and Match-Box focus on ungapped alignments across segments, utilizing thresholds for optimal alignment.Despite automation, alignments may need manual editing to correct misalignments and improve biological relevance, emphasizing empirics in adjustments.
SCORING FUNCTION
Multiple sequencealignment is to arrange sequences in
such a way that a maximum number of residues from each
sequence are matched up according to a particular scoring
function.
The scoring function for multiple sequence alignment is
based on the concept of sum of pairs (SP)
In calculating the SP scores, each column is scored by
summing the scores for all possible pairwise matches,
mismatches and gap costs.
SEQUENCE 1 G K N
SEQUENCE 2 T R N
SEQUENCE 3 S H E
SUM OF PAIRS -2 +1 +6 = 5
4.
The scoring isbased on the BLOSUM62 matrix . The total score for
the alignment is 5, which means that the alignment is 25 = 32
times more likely to occur among homologous sequences than by
random chance.
EXHAUSTIVE ALGORITHMS
The exhaustive alignment method involves examining all possible
aligned positions simultaneously.
Similar to dynamic programming in pairwise alignment, which involves
the use of a two-dimensional matrix to search for an optimal
alignment, to use dynamic programming for multiple sequence
alignment,
extra dimensions are needed to take all possible ways of sequence
matching into consideration.
DCA (Divide-and-Conquer Alignment, http://bibiserv.techfak.uni-
bielefeld.de/ dca/) is a web-based program that is in fact
semiexhaustive because certain steps of computation are reduced to
heuristics(LEARN NEW SOMETHING )
5.
can handle onlydatasets of a very limited number of sequences.
HEURISTIC ALGORITHMS
• The use of dynamic programming is not feasible for routine
multiple sequence alignment, faster and heuristic algorithms
have been developed. The heuristic algorithms fall into three
categories:
• 1. Progressive alignment type
• 2. Iterative alignment type,
• 3. Block-based alignment type.
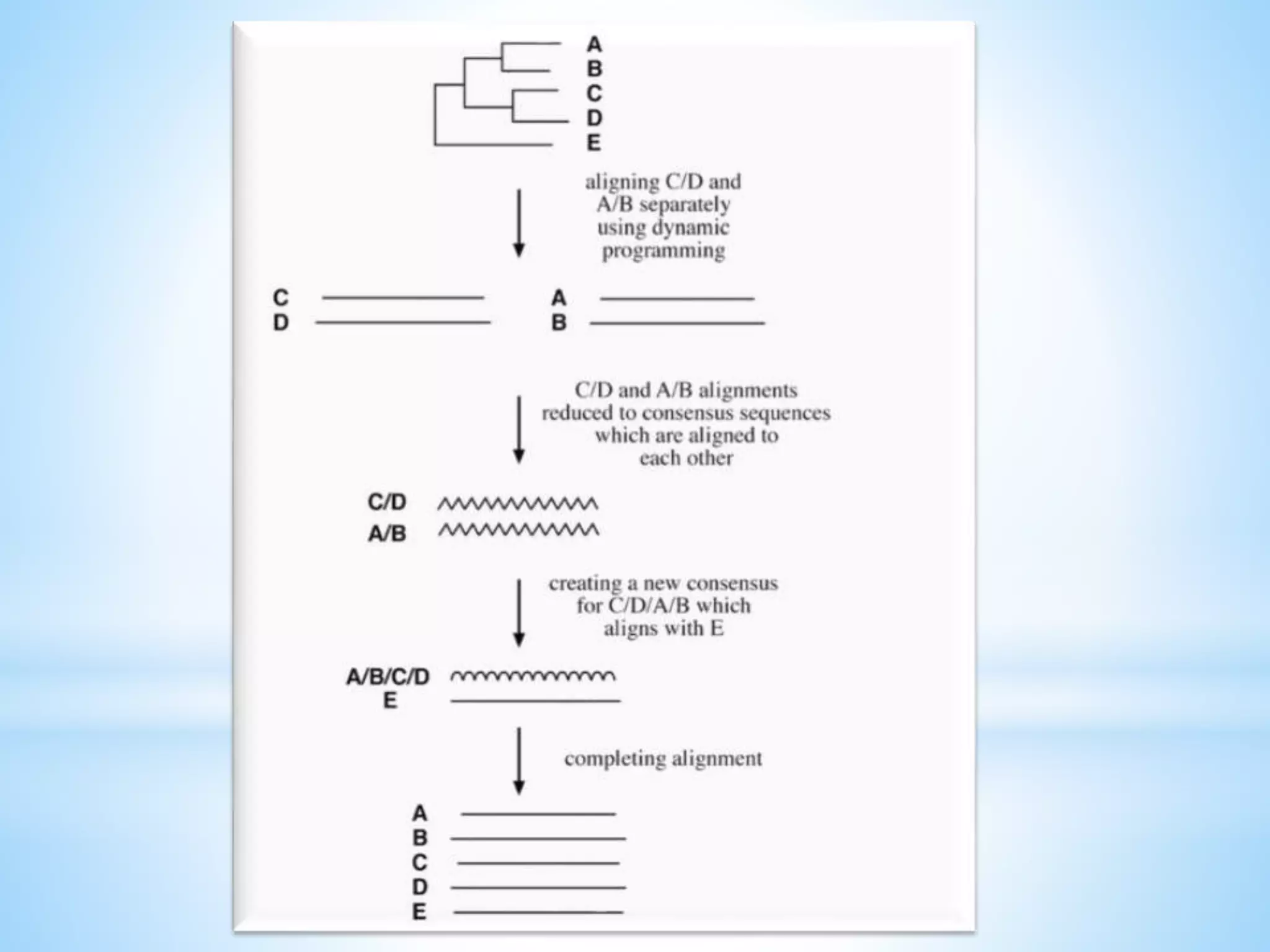
Progressive Alignment Method:
Progressive alignment depends on the stepwise assembly of
multiple alignment and is heuristic in nature
It first conducts pairwise alignments for each possible pair of
sequences using the Needleman–Wunsch global alignment method
and records these similarity scores from the pairwise comparisons
6.
The scores caneither be percent identity or similarity scores
based on a particular substitution matrix
Probably the most well-known progressive alignment program is
Clustal.
Some of its important features are introduced next. Clustal
(www.ebi.ac.uk/clustalw/) is a progressive multiple alignment
program available either as a stand-alone or on-line program.
The stand-alone program, which runs on UNIX and Macintosh, has
two variants, ClustalW and ClustalX.
9.
HEURISTIC ALGORITHMS
Clustal doesnot rely on a single substitution matrix. Instead, it
applies different scoring matrices when aligning sequences,
depending on degrees of similarity
For example, for closely related sequences that are aligned in the
initial steps, Clustal automatically uses the BLOSUM62 or PAM120
matrix. When more divergent sequences are aligned in later steps
of the progressive alignment, the BLOSUM45 or PAM250 matrices
may be used instead.
Another feature of Clustal is the use of adjustable gap penalties
that allow more insertions and deletions in regions that are
outside the conserved domains, but fewer in conserved regions.
The program also applies a weighting scheme to increase the
reliability of aligning divergent sequences (sequences with less
than 25% identity)
Drawbacks and Solutions
10.
T-Coffee (Tree-based ConsistencyObjective Function for alignment
Evaluation; www.ch.embnet.org/software/TCoffee.html) performs
progressive sequence alignments as in Clustal.
Because an optimal initial alignment is chosen from many alternative
alignments, T-Coffee avoids the problem of getting stuck in the
suboptimal alignment regions, which minimizes errors in the early
stages of alignment assembly.
Poa (Partial order alignments, www.bioinformatics.ucla.edu/poa/) is
a progressive alignment program that does not rely on guide trees.
Instead, the multiple alignment is assembled by adding sequences in
the order they are given
12.
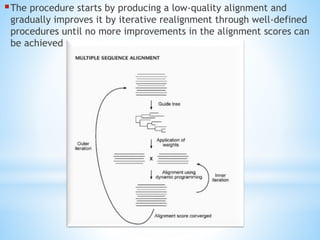
The procedure startsby producing a low-quality alignment and
gradually improves it by iterative realignment through well-defined
procedures until no more improvements in the alignment scores can
be achieved
13.
Block-Based Alignment
• Theprogressive and iterative alignment strategies are largely
global alignment based and may therefore fail to recognize
conserved domains and motifs
• The strategy identifies a block of ungapped alignment shared by
all the sequences, hence, the block-based local alignment
strategy. Two block-based alignment programs
DIALIGN2
(http://bioweb.pasteur.fr/seqanal/interfaces/dialign2.html) is a
webbased program
The method breaks each of the sequences down to smaller
segments and performs all possible pairwise alignments between
the segments. High-scoring segments, called blocks,
•Match-Box
• (www.sciences.fundp.ac.be/biologie/bms/matchbox
submit.shtml) is a web-based server
14.
• The programcompares segments of every nine residues of all possible
pairwise alignments. If the similarity of particular segments is above a
certain threshold across all sequences, they are used as an anchor to
assemble multiple alignments; residues between blocks are unaligned.
Protein-Coding DNA Sequences
1. The process of achieving maximum sequence similarity at the DNA
level, mismatches of genetic codons occur that violate the accepted
evolutionary scenario that insertions or deletions occur in units of
codons.
2. E.G errors can occur when two sequences are being compared at the
protein and DNA levels
Editing
No matter how good an alignment program seems, the automated
alignment often contains misaligned regions
This involves introducing or removing gaps to maximize biologically
meaningful matches. Sometimes, portions that are ambiguously aligned
and deemed to be incorrect have to deleted. In manual editing,
empirical evidence or mere experience is needed to make corrections
on an alignment.