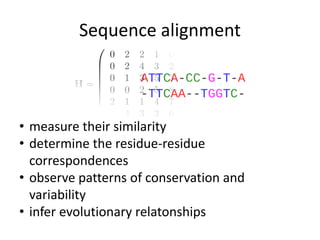
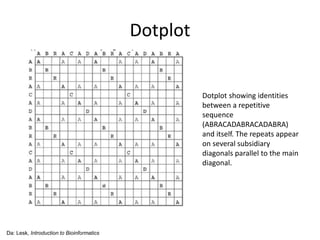
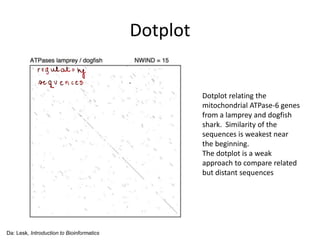
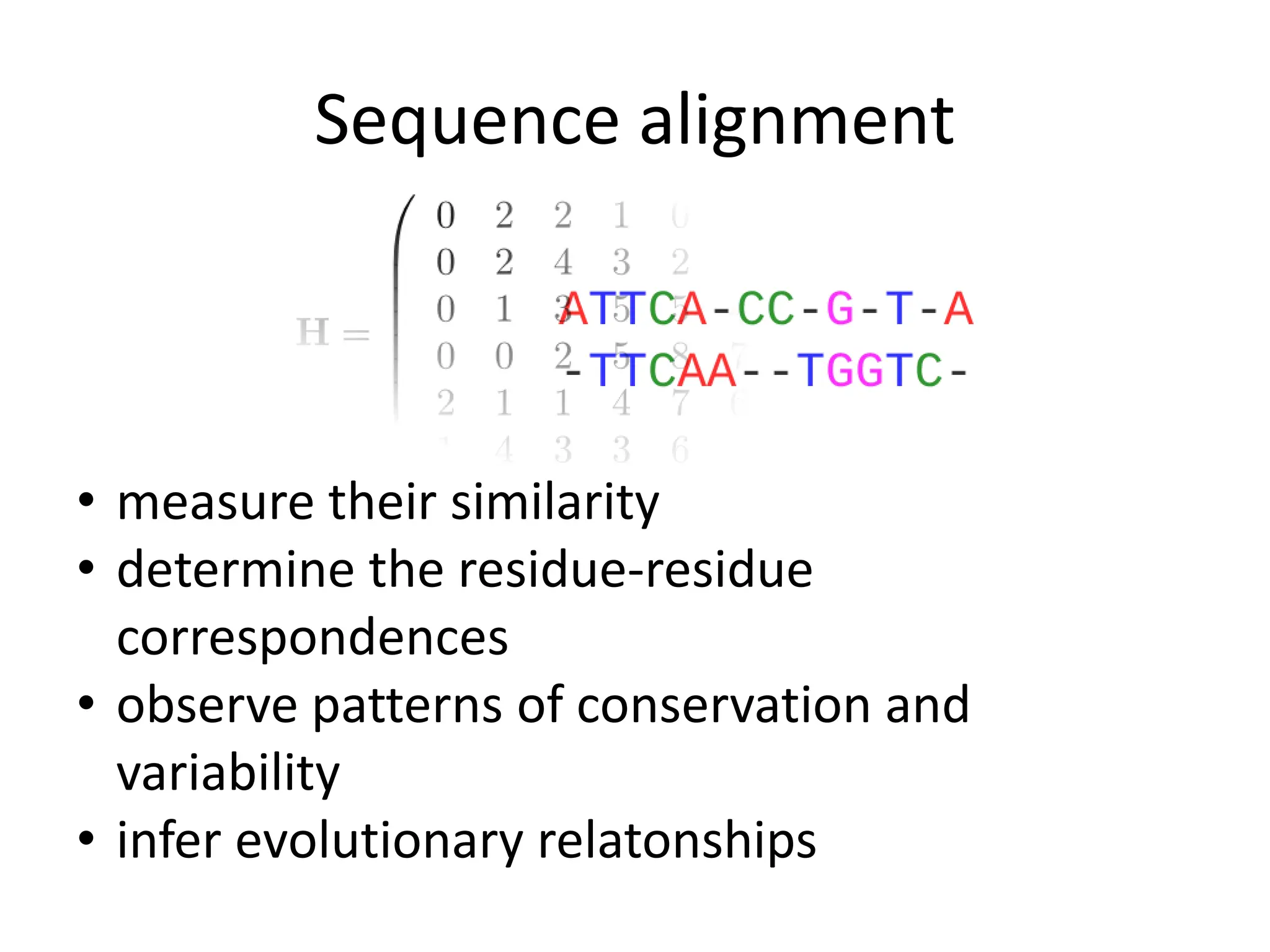
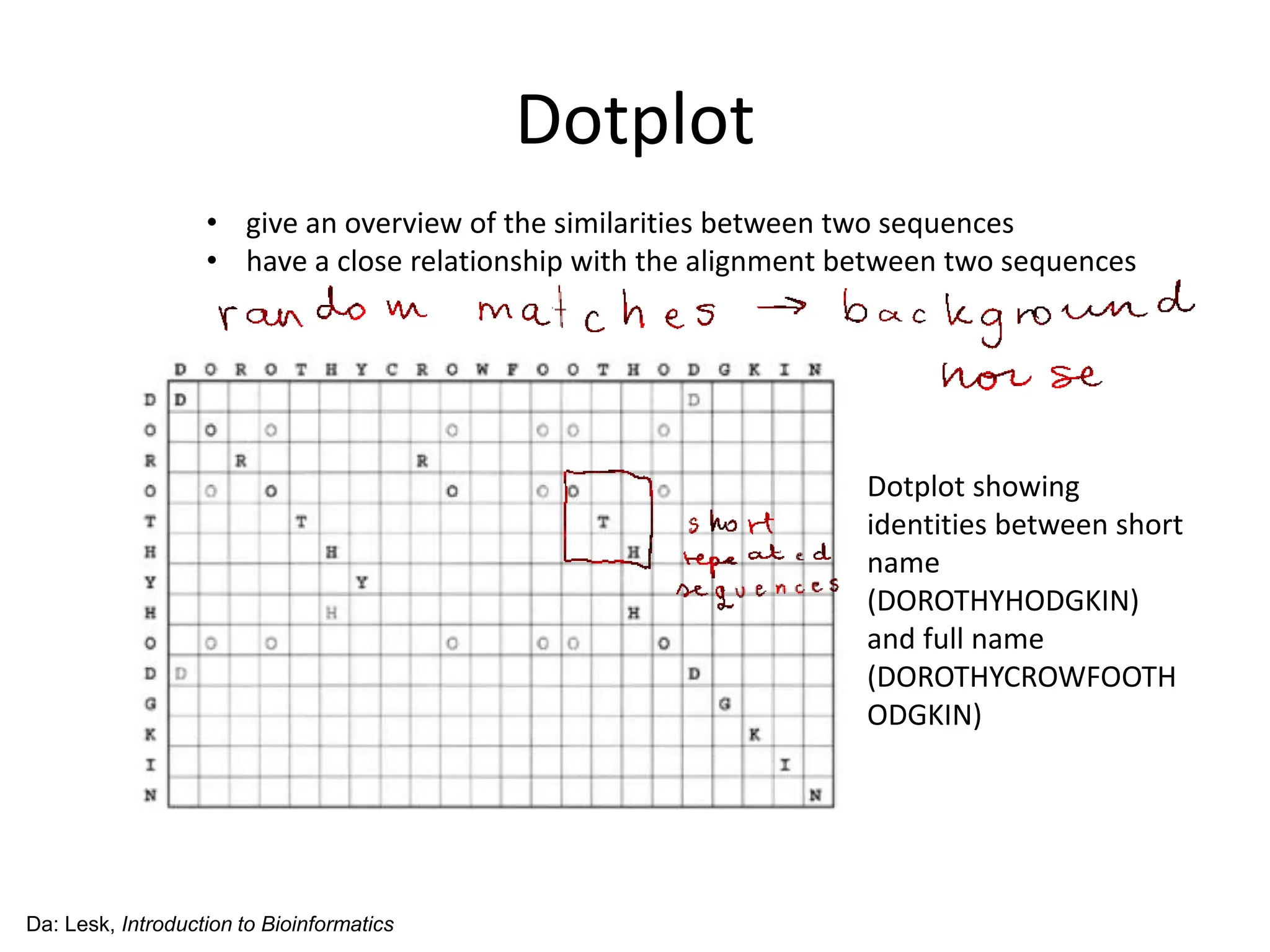
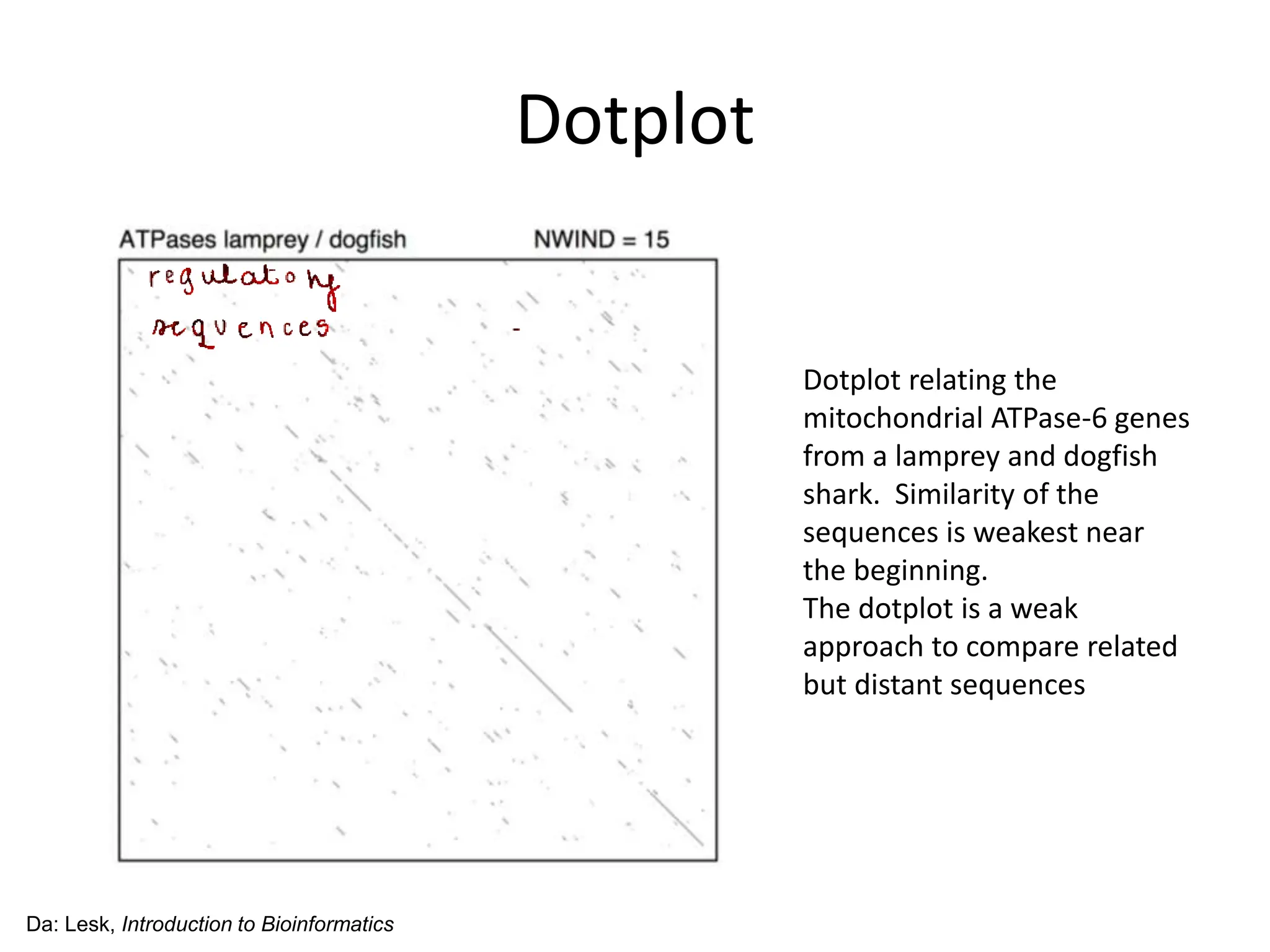
The document discusses various methods for sequence alignment in bioinformatics, focusing on measuring similarity and establishing residue-residue correspondences. It highlights the importance of algorithms in identifying optimal alignments and introduces tools like dotplots to visualize sequence similarities. Additionally, it defines distance measures such as Hamming and Levenshtein distances to compare sequences of equal and varying lengths.