Downloaded 60 times





































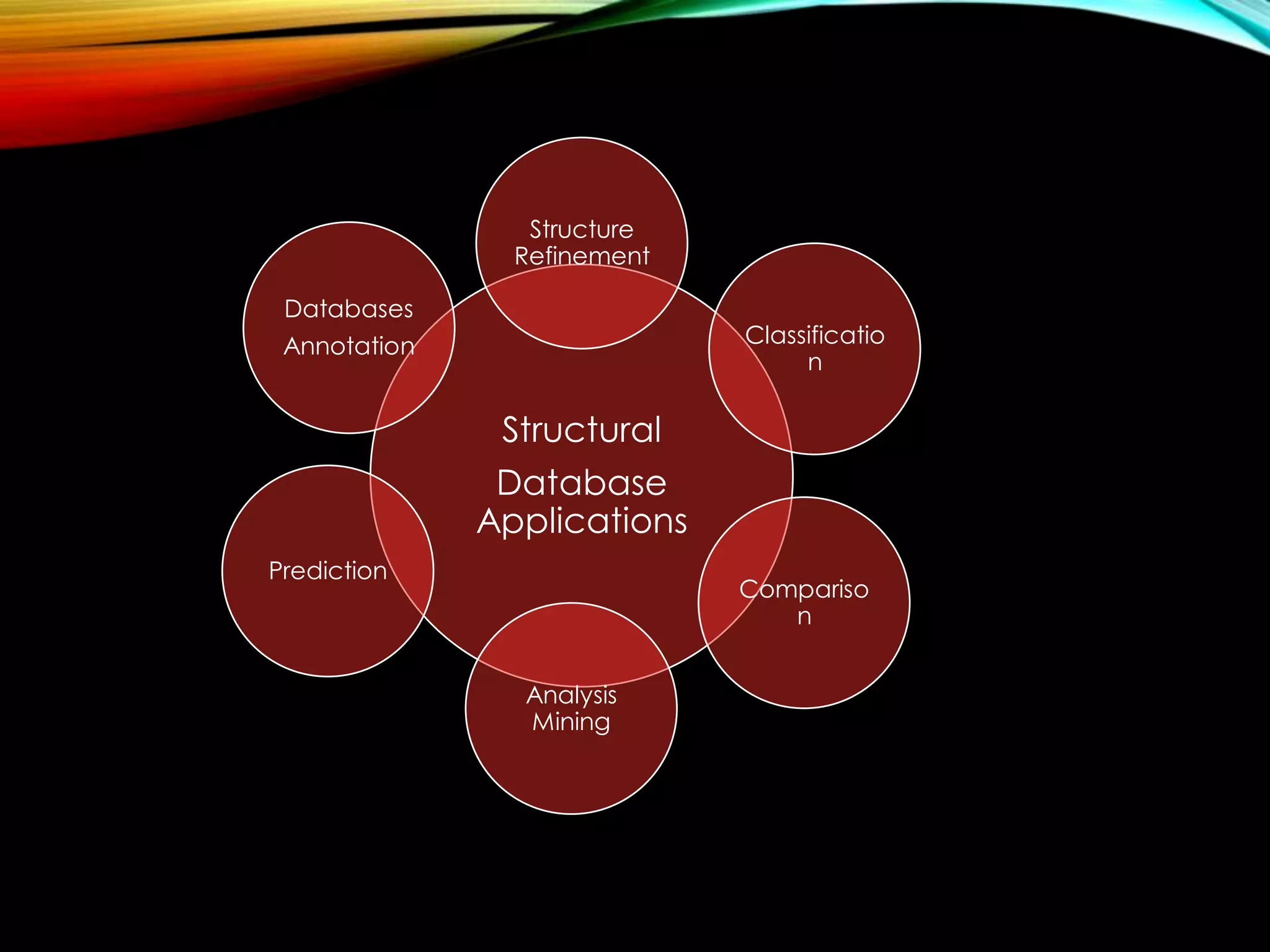


Structural databases like PDB, CSD, and CATH contain 3D structural information of proteins, small molecules, and macromolecules determined through techniques like X-ray crystallography and NMR spectroscopy. These databases provide bibliographic data, atomic coordinates, and other details for each entry. PDB contains protein structures, CSD contains organic and metal-organic structures, and CATH classifies protein domains hierarchically. Structural databases have wide applications in structure prediction, analysis, mining, comparison, classification, structure refinement, and database annotation.
Introduction to structural databases like PDB, CSD, and CATH, their purpose in crystallography, and common information they hold.
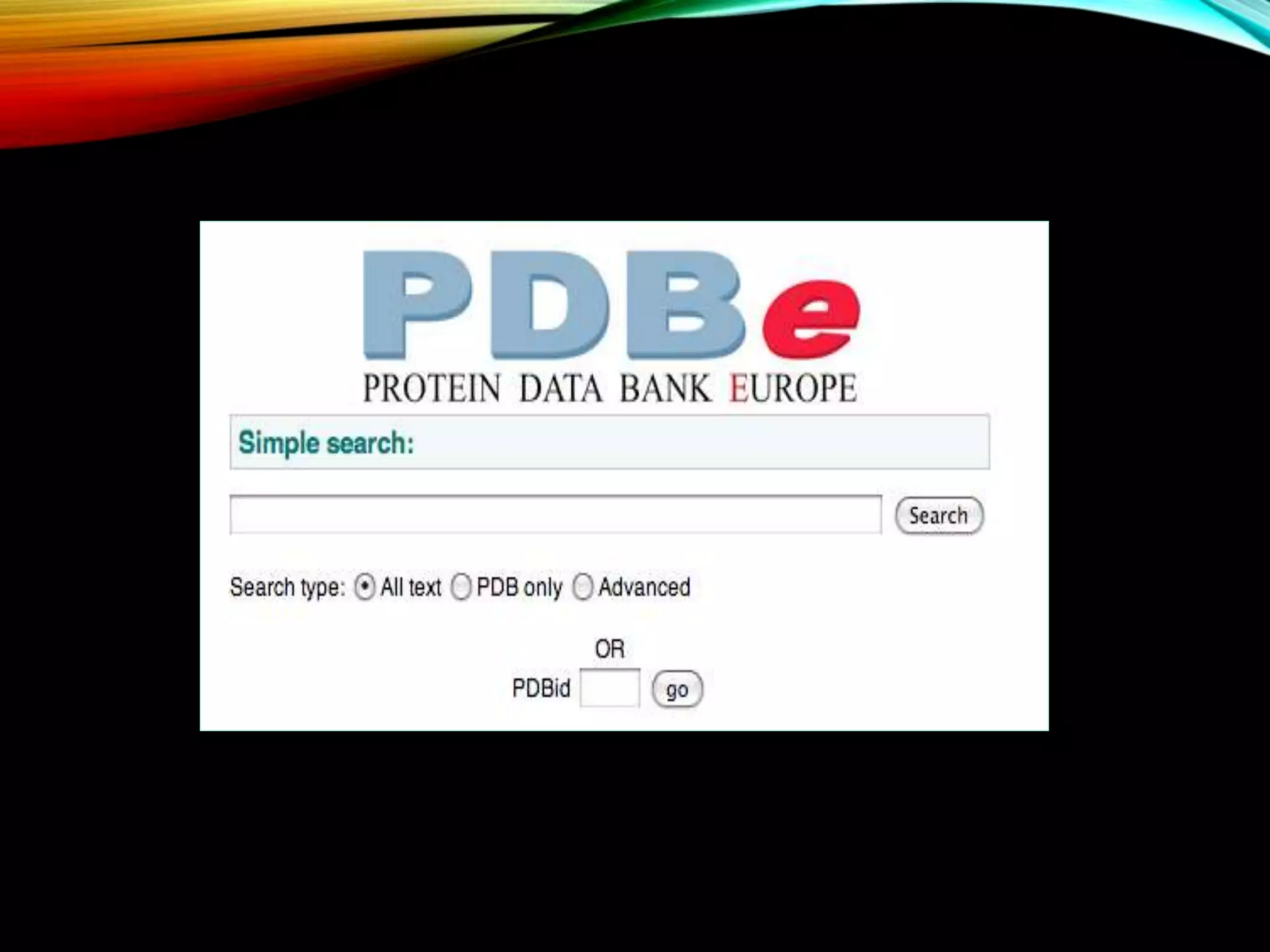
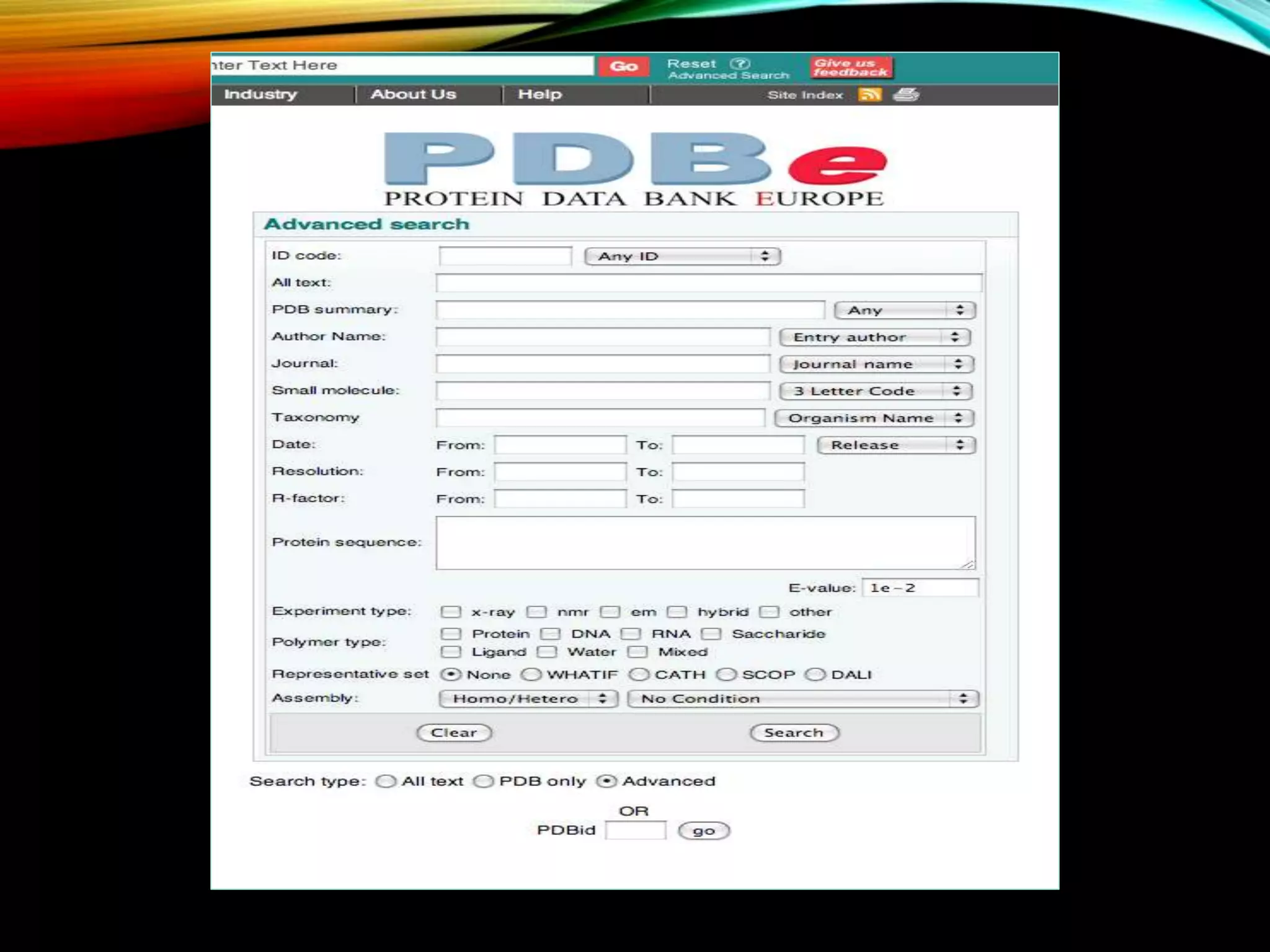
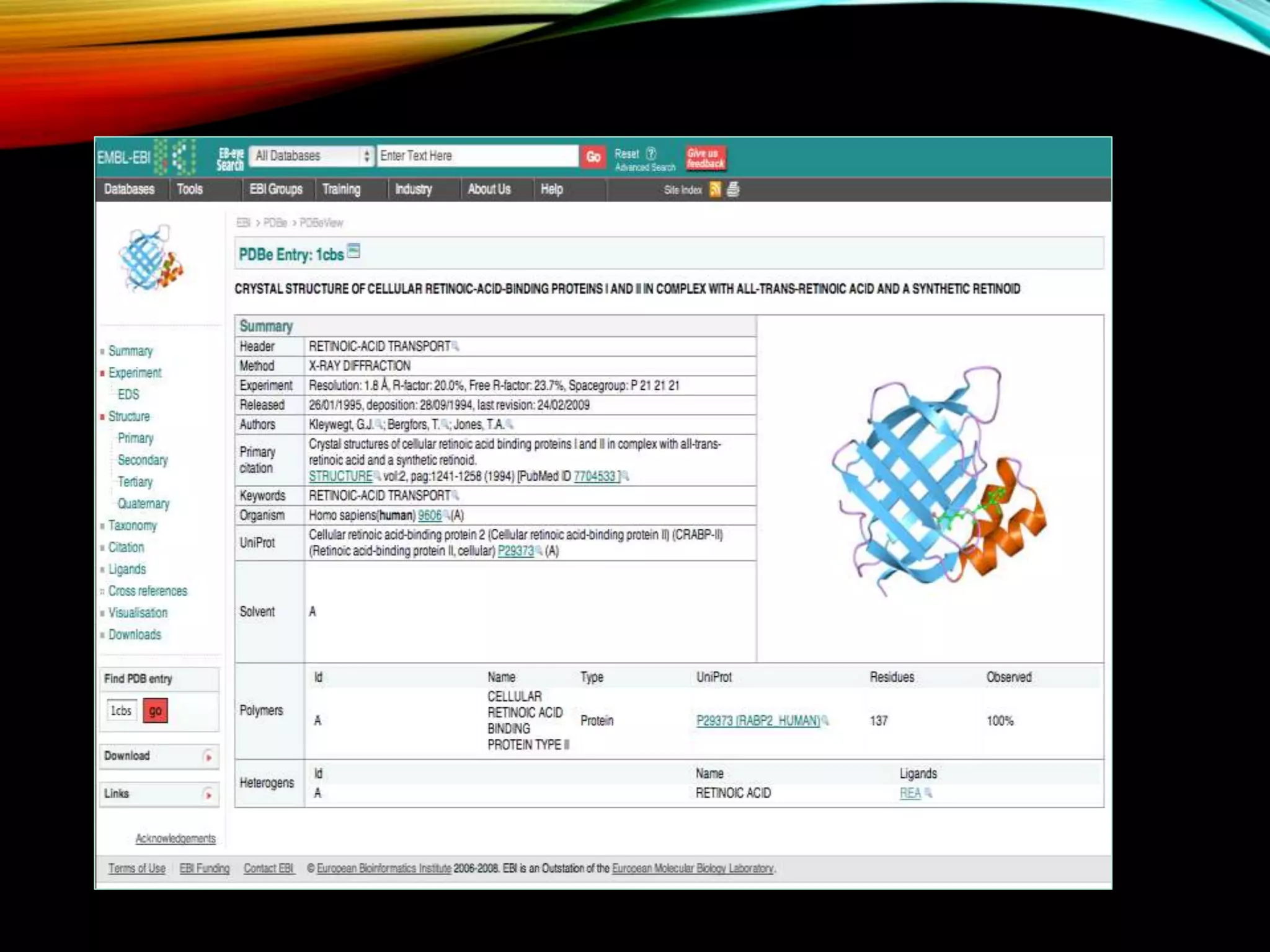
Details about PDB, its functions, and management by wwPDB, containing 3D protein structures obtained via various techniques.
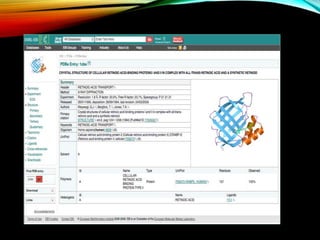
Description of PDB file format including records like HEADER, TITLE, COMPND, ATOM, and their informational significance.
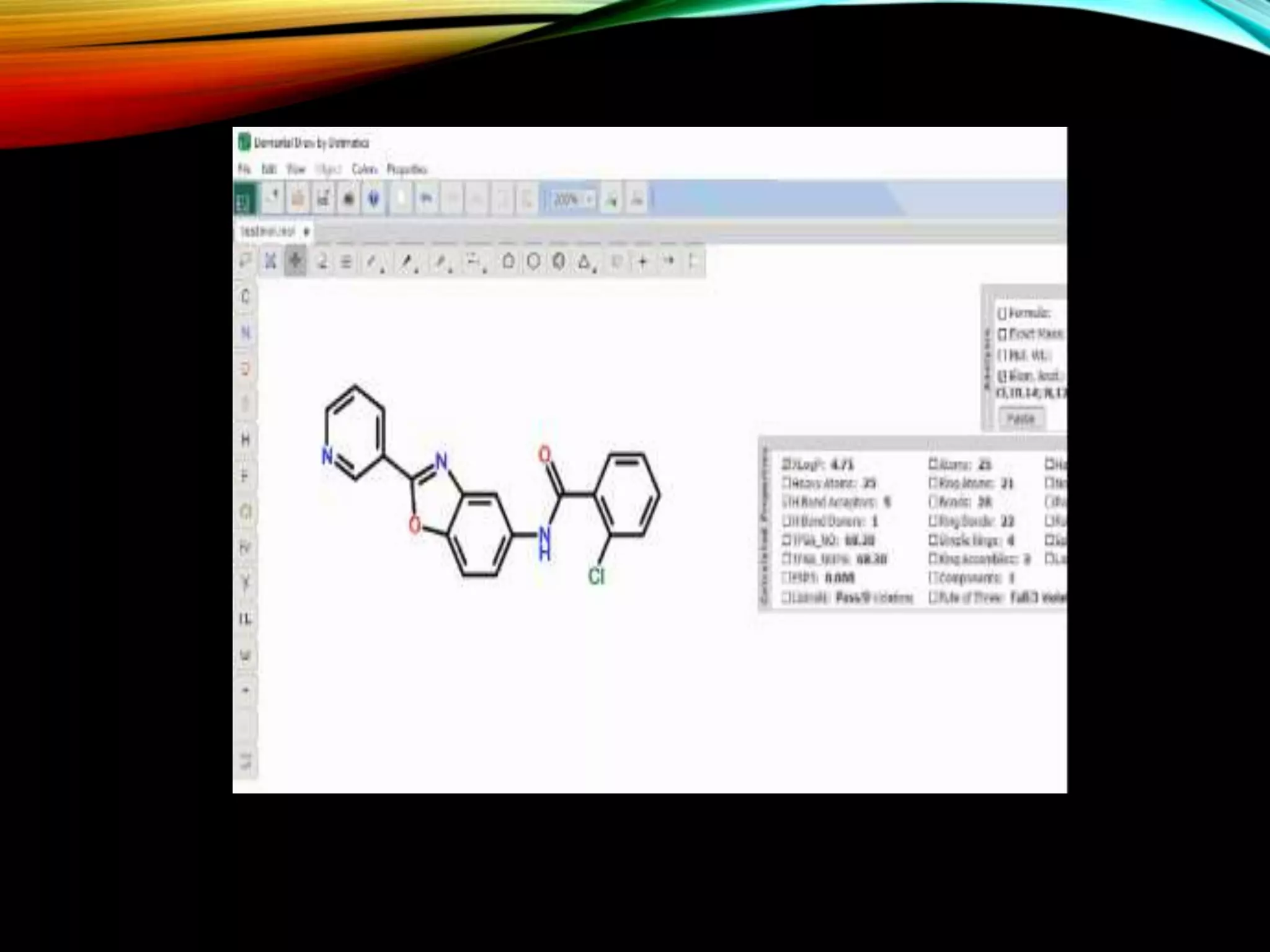
An example of a PDB file format illustrating how structural information is presented in a structured manner.
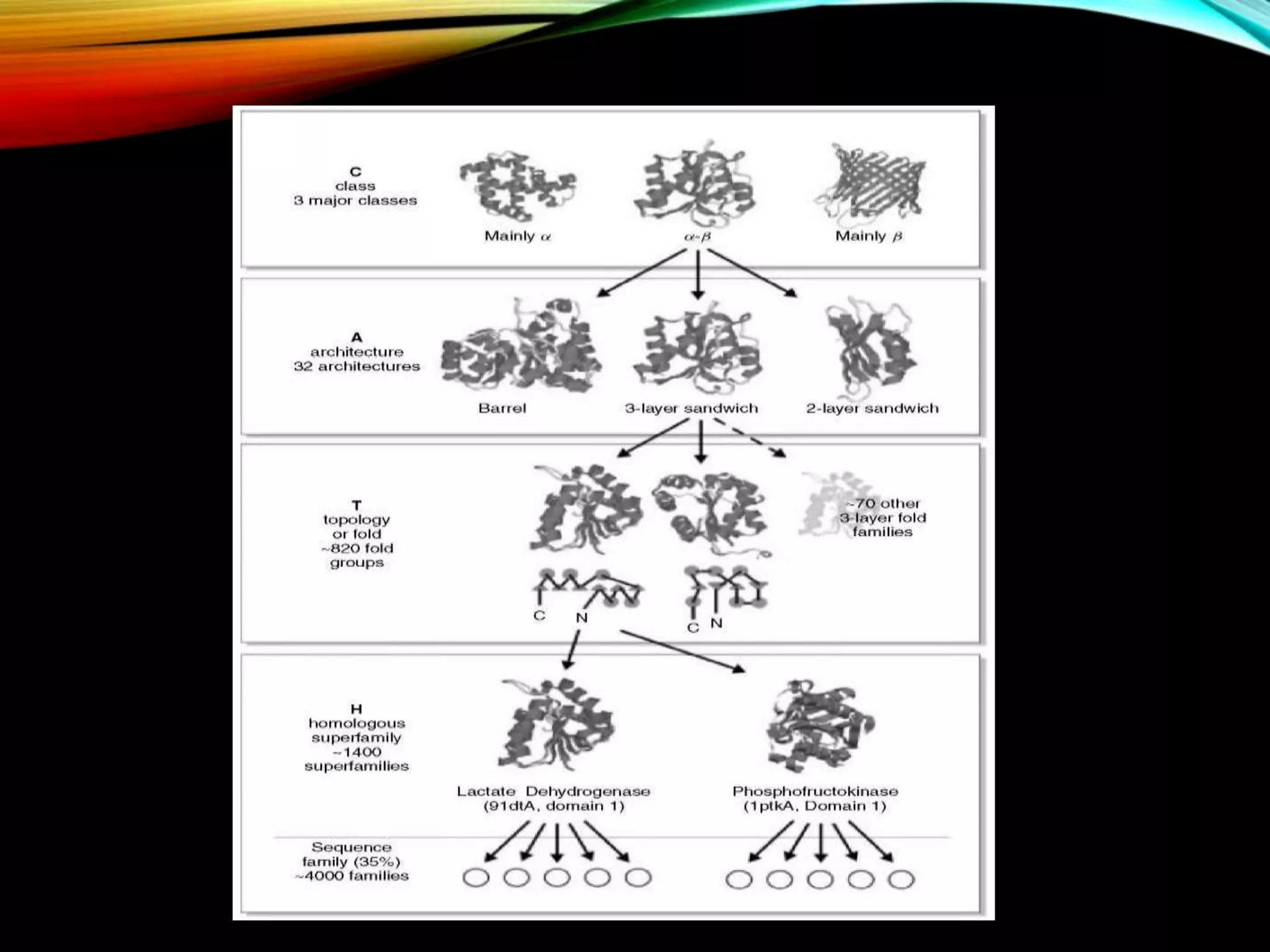
Introduction to CATH focusing on protein classification, its levels, and their significance in understanding protein structures.
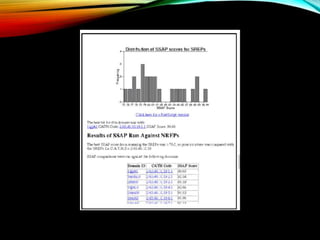
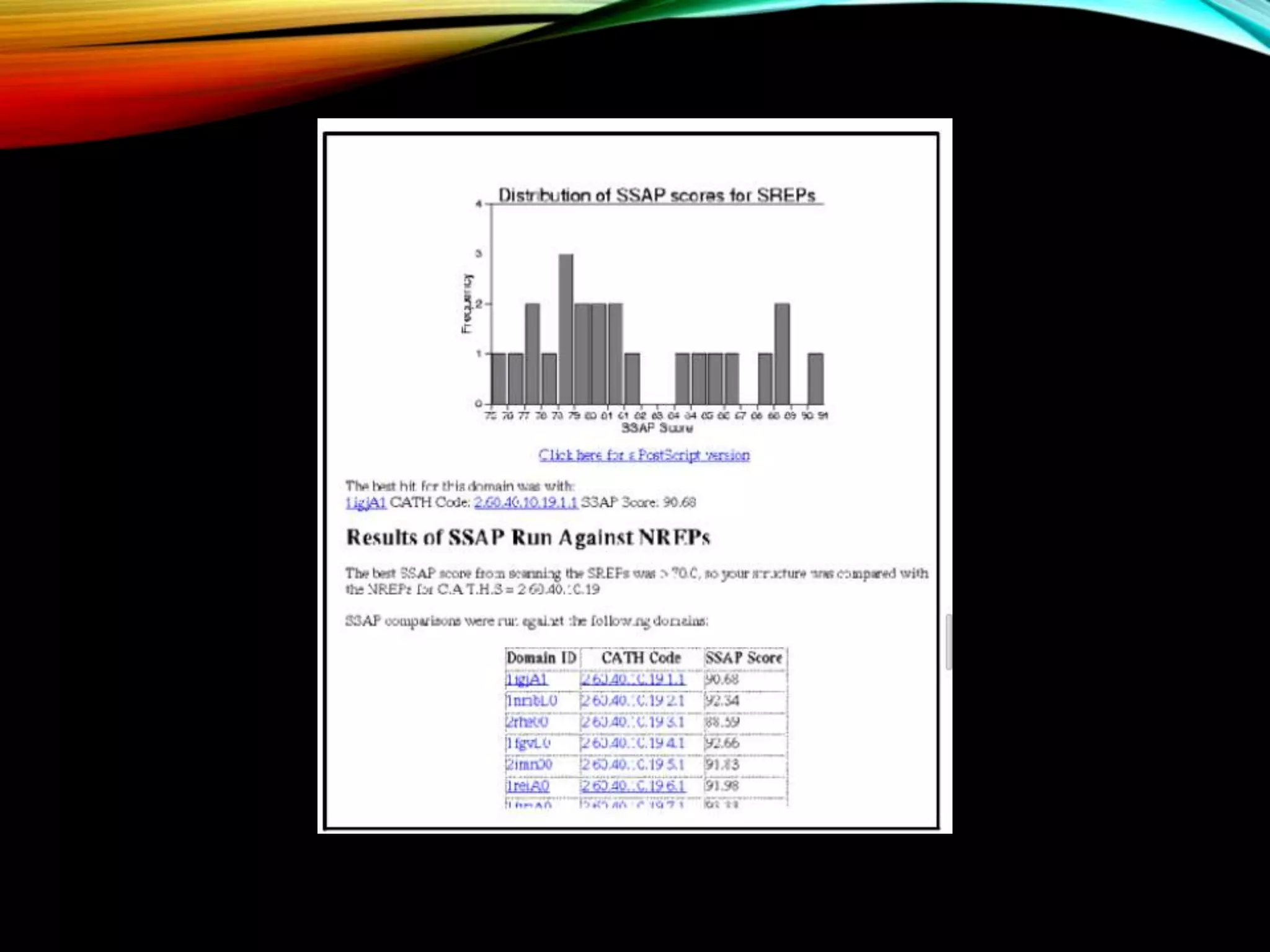
CATH database aims and features including structural classifications, domain submission, and detection programs for protein analysis.
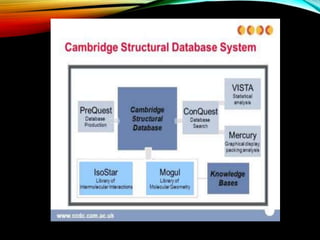
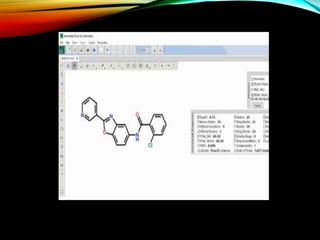
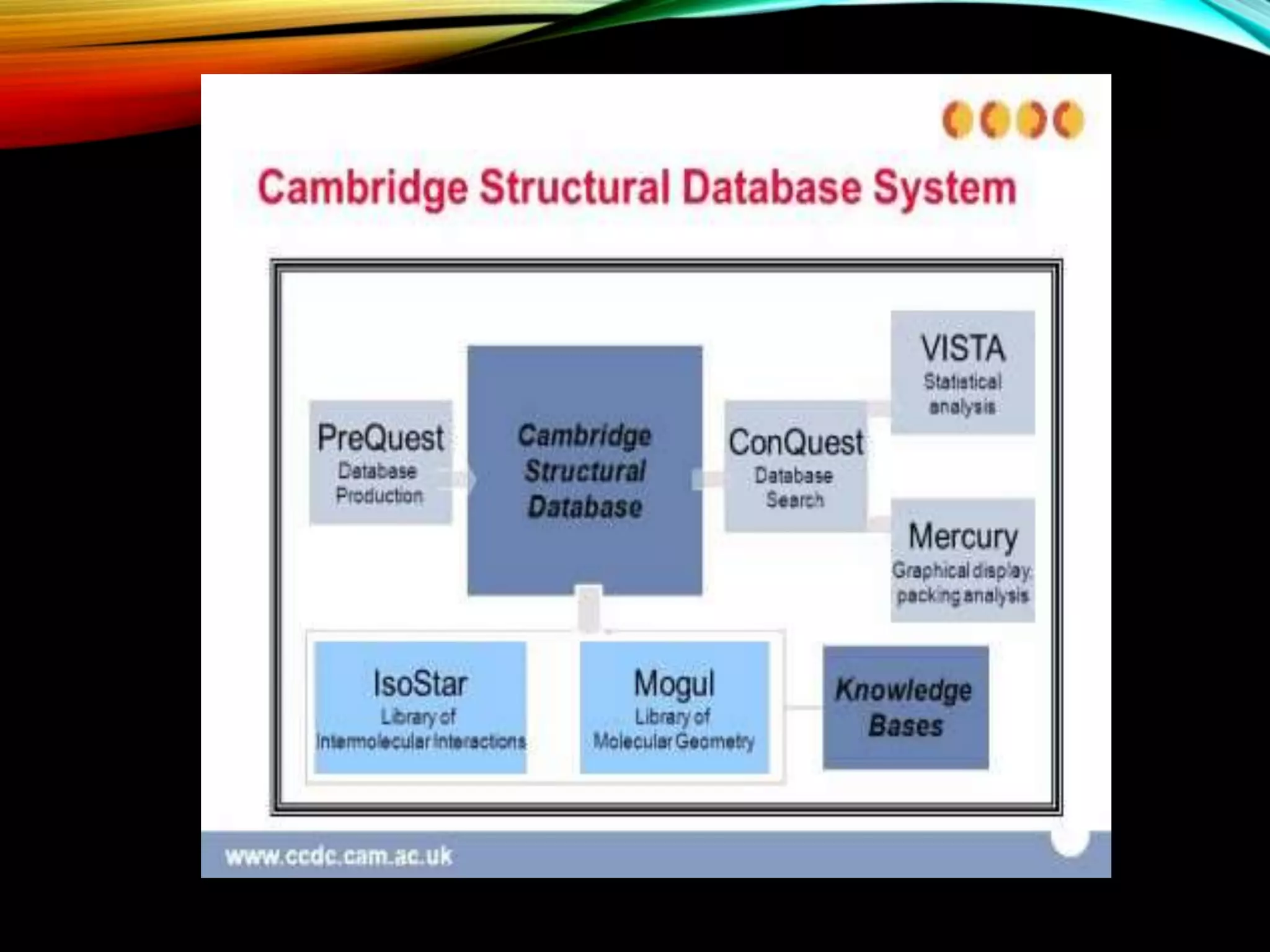
Description of CSD as a resource for 3D structures of organic compounds, its maintenance, and contribution by researchers globally.
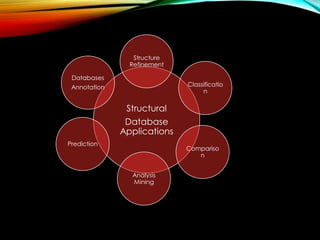
Discussion on different applications of structural databases such as prediction, analysis, mining, comparison, and classification.

















































































