The document provides an overview of sequence alignment, including:
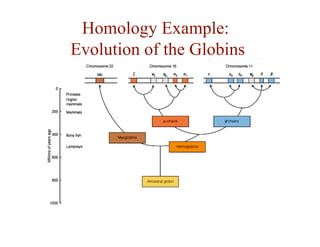
- The task of sequence alignment is to determine the correspondences between substrings in sequences that maximize a similarity score.
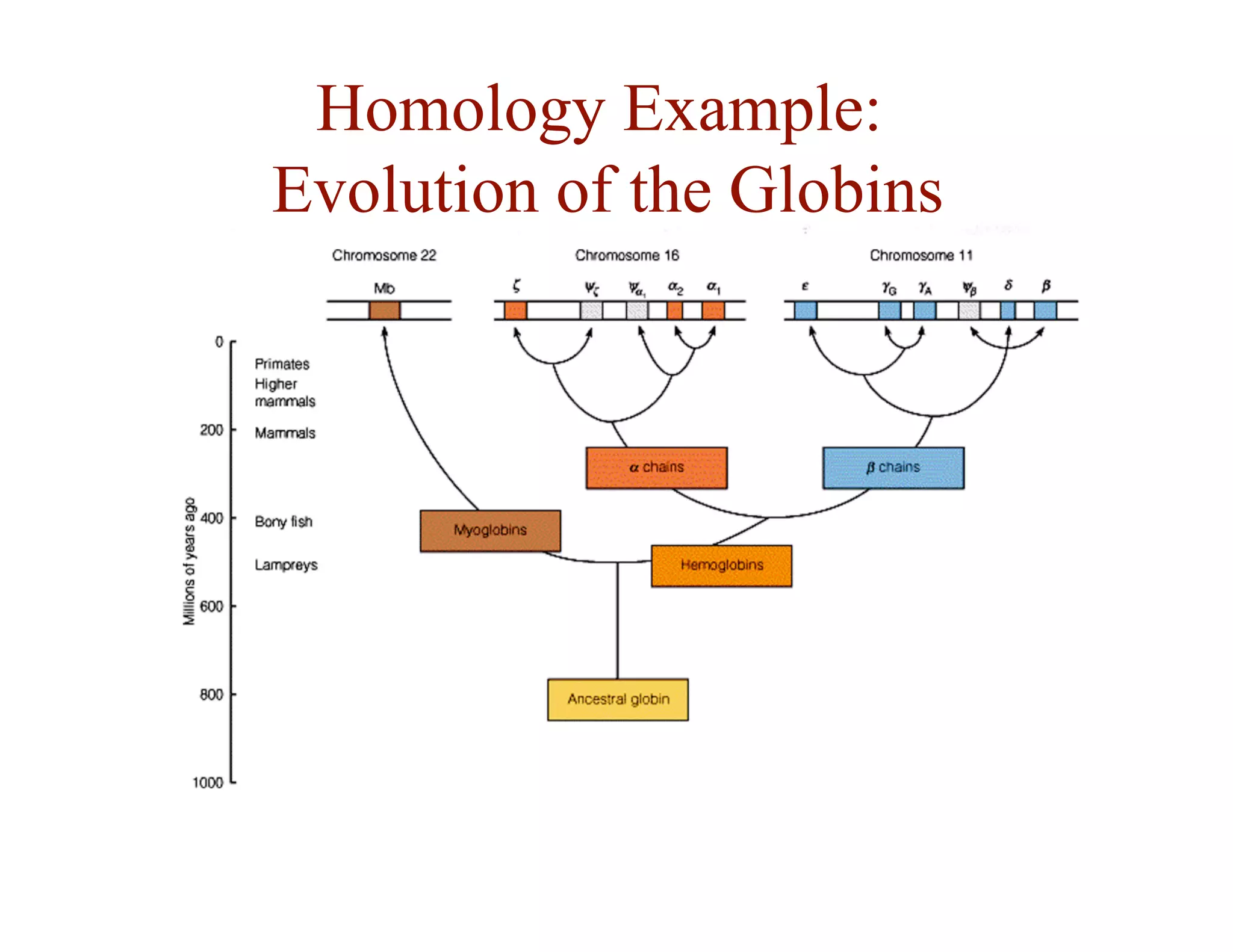
- Alignment allows inference of homology and function based on sequence similarity.
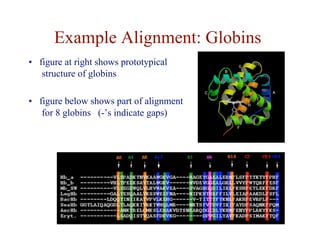
- Key issues include variable sequence lengths, small matching regions, and modeling substitutions and gaps.
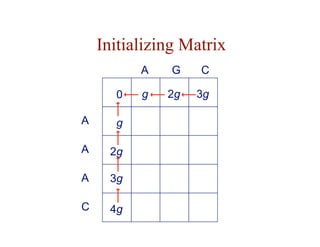
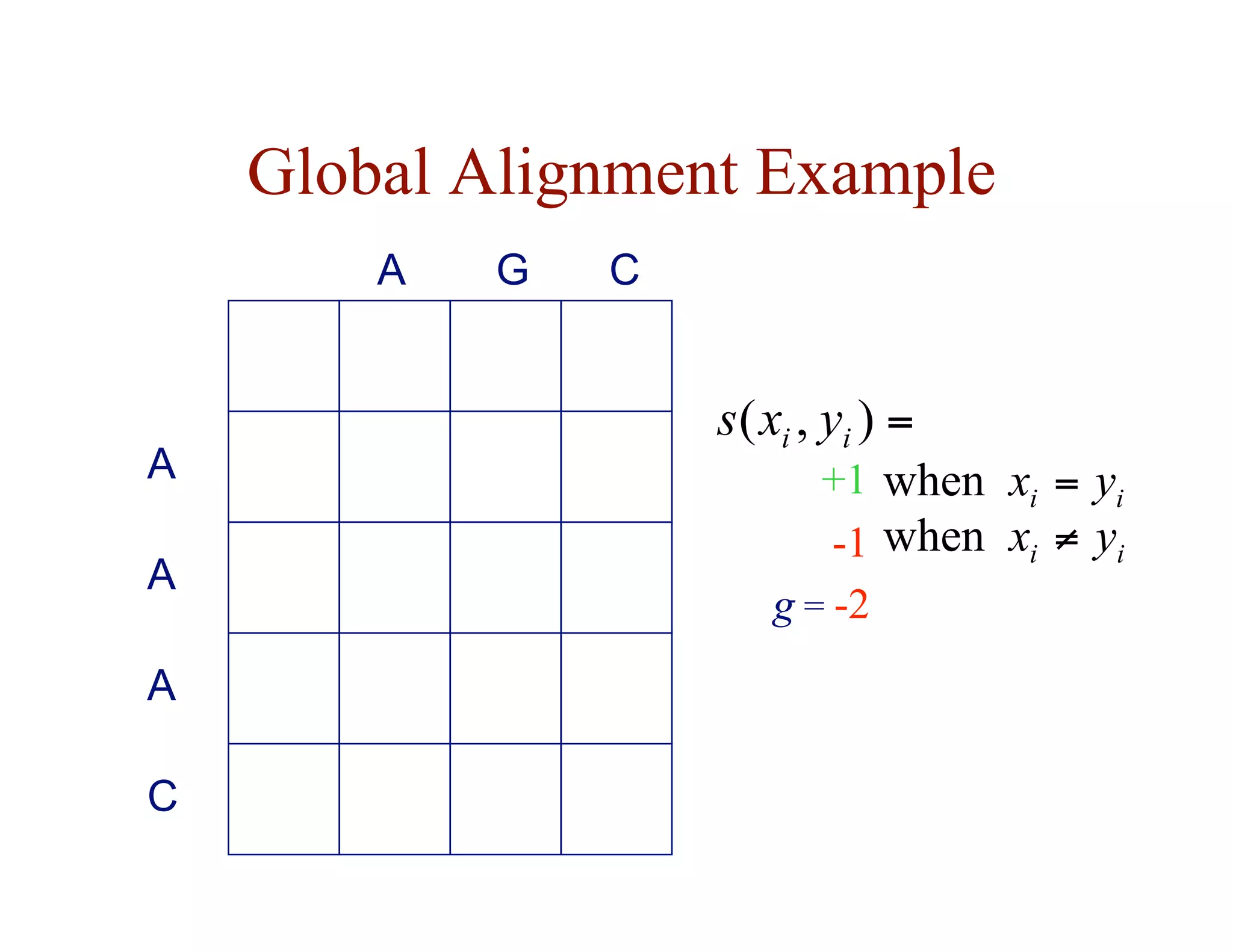
- Dynamic programming is used to find optimal global and local alignments in quadratic time by solving subproblems and reusing results.















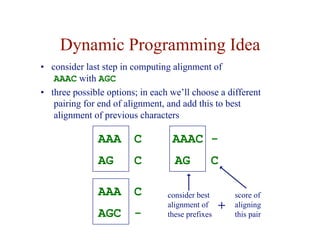
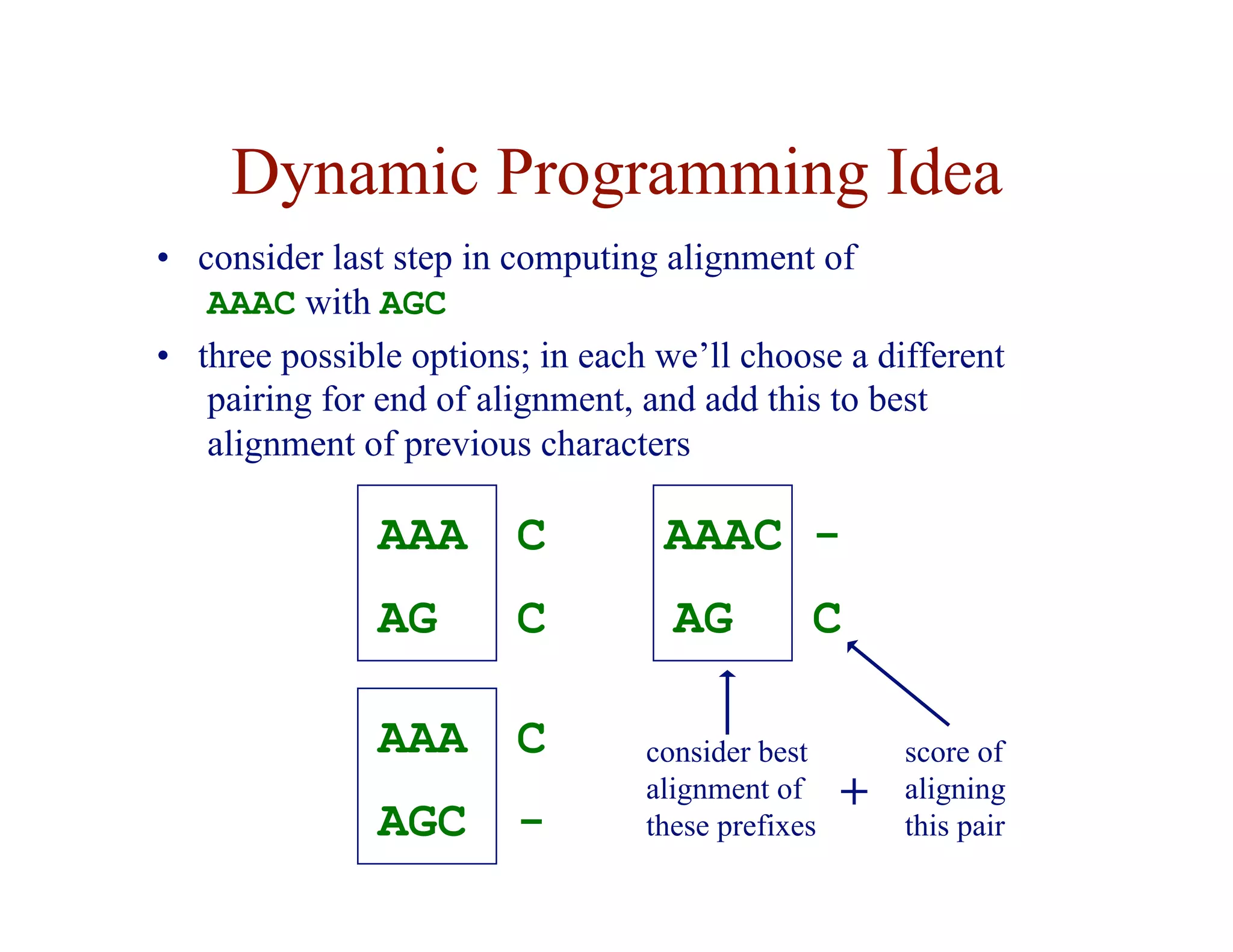
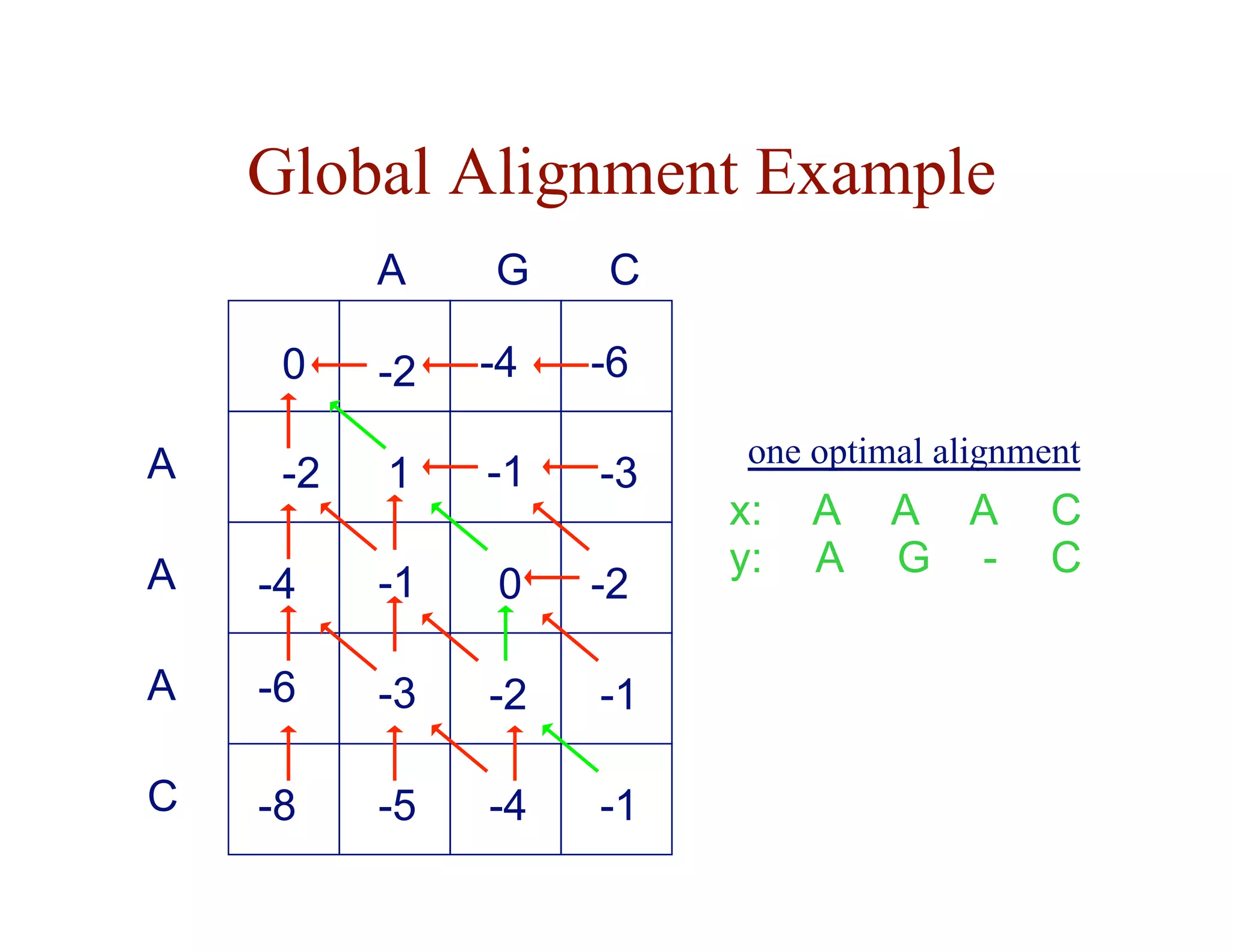
![Dynamic Programming Idea
• given an n-character sequence x, and an m-character
sequence y
• construct an (n+1) × (m+1) matrix F
• F ( i, j ) = score of the best alignment of
x[1…i ] with y[1…j ]
A
A
C
A G
A
C
score of best alignment of
AAA to AG](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/85/02-alignment-pdf-19-320.jpg)



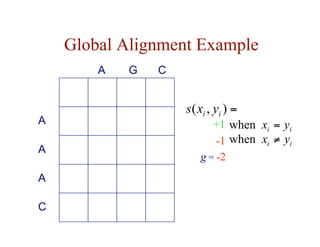
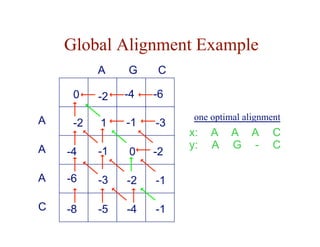

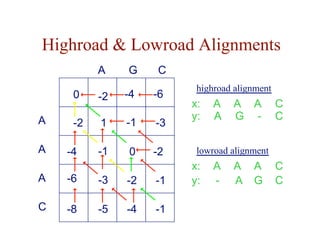



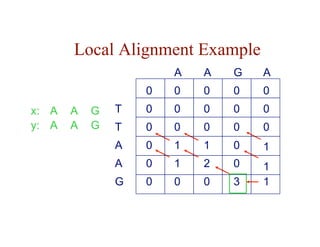
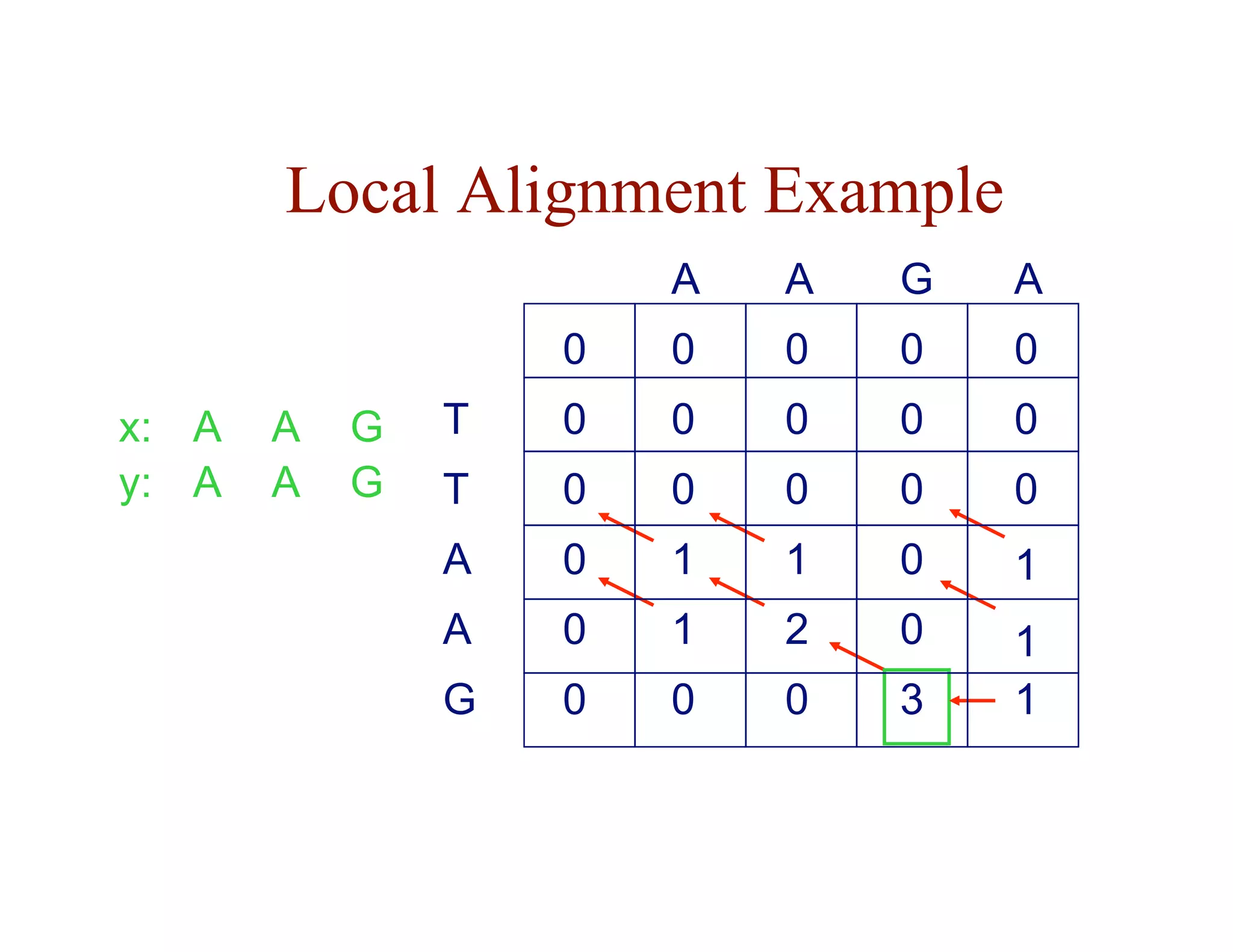
![Local Alignment DP Algorithm
• original formulation: Smith & Waterman, Journal of
Molecular Biology, 1981
• interpretation of array values is somewhat different
– F ( i, j ) = score of the best alignment of a suffix of
x[1…i ] and a suffix of y[1…j ]](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/85/02-alignment-pdf-31-320.jpg)





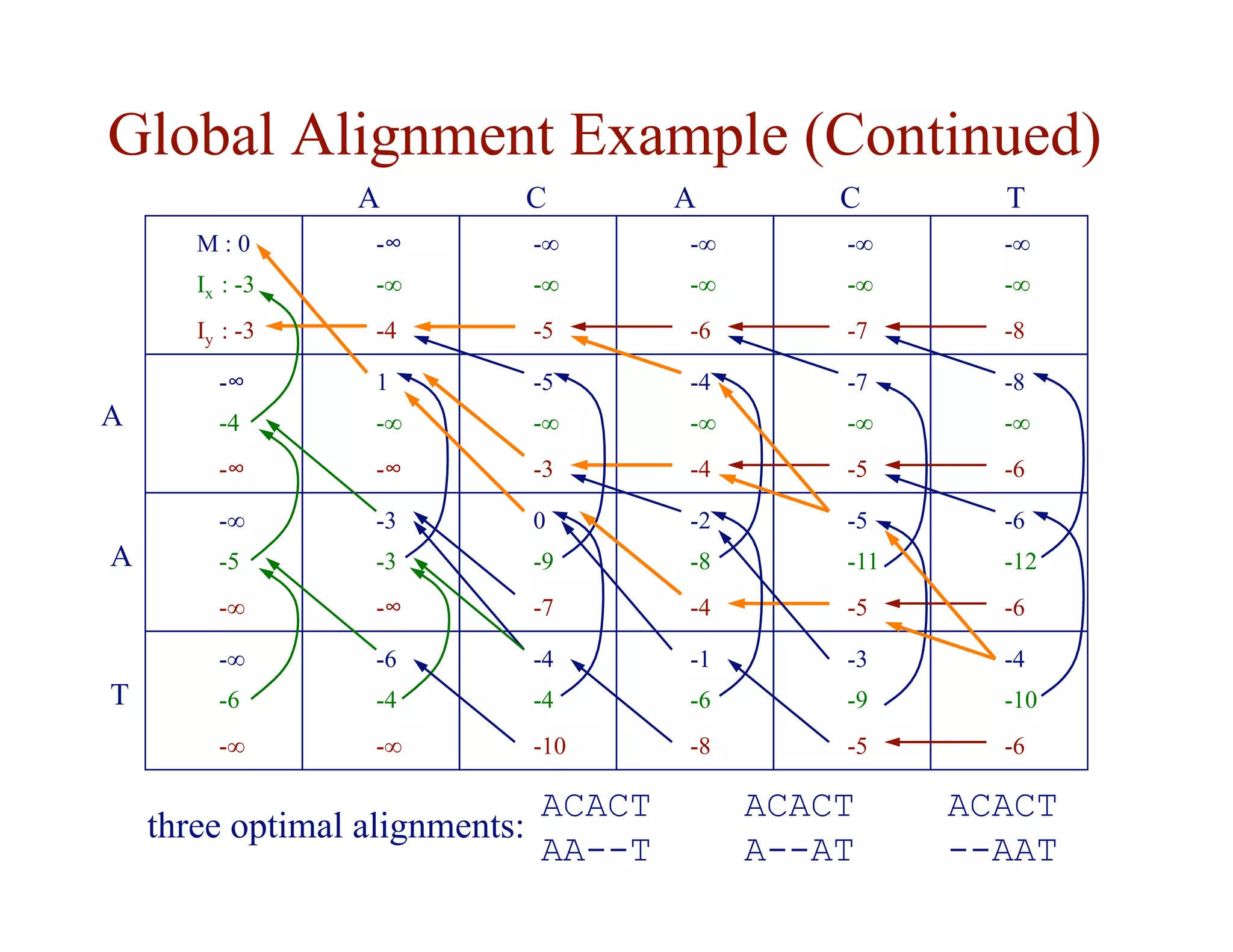
![Dynamic Programming for the
Affine Gap Penalty Case
• to do in time, need 3 matrices instead of 1
)
,
( j
i
M
)
,
( j
i
Ix
)
,
( j
i
Iy
best score given that y[j] is
aligned to a gap
best score given that x[i] is
aligned to a gap
best score given that x[i] is
aligned to y[j]
)
( 2
n
O](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/85/02-alignment-pdf-38-320.jpg)







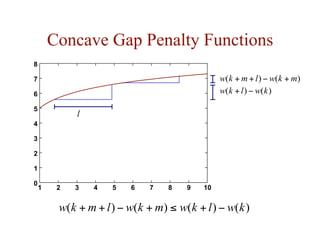

![Substitution Matrices
• two popular sets of matrices for protein sequences
– PAM matrices [Dayhoff et al., 1978]
– BLOSUM matrices [Henikoff & Henikoff, 1992]
• both try to capture the the relative substitutability of amino
acid pairs in the context of evolution](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/85/02-alignment-pdf-48-320.jpg)

![Heuristic Methods
• the algorithms we learned today take O(nm) time to align
sequences, which is too slow for searching large databases
– imagine an internet search engine, but where queries and results
are protein sequences
• heuristic methods do fast approximation to dynamic
programming
– example: BLAST [Altschul et al., 1990; Altschul et al., 1997]
– break sequence into small (e.g. 3 base pair) “words”
– scan database for word matches
– extend all matches to seek high-scoring alignments
– tradeoff: sensitivity for speed](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/85/02-alignment-pdf-50-320.jpg)


















![Dynamic Programming Idea
• given an n-character sequence x, and an m-character
sequence y
• construct an (n+1) × (m+1) matrix F
• F ( i, j ) = score of the best alignment of
x[1…i ] with y[1…j ]
A
A
C
A G
A
C
score of best alignment of
AAA to AG](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/75/02-alignment-pdf-19-2048.jpg)








![Local Alignment DP Algorithm
• original formulation: Smith & Waterman, Journal of
Molecular Biology, 1981
• interpretation of array values is somewhat different
– F ( i, j ) = score of the best alignment of a suffix of
x[1…i ] and a suffix of y[1…j ]](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/75/02-alignment-pdf-31-2048.jpg)





![Dynamic Programming for the
Affine Gap Penalty Case
• to do in time, need 3 matrices instead of 1
)
,
( j
i
M
)
,
( j
i
Ix
)
,
( j
i
Iy
best score given that y[j] is
aligned to a gap
best score given that x[i] is
aligned to a gap
best score given that x[i] is
aligned to y[j]
)
( 2
n
O](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/75/02-alignment-pdf-38-2048.jpg)







![Substitution Matrices
• two popular sets of matrices for protein sequences
– PAM matrices [Dayhoff et al., 1978]
– BLOSUM matrices [Henikoff & Henikoff, 1992]
• both try to capture the the relative substitutability of amino
acid pairs in the context of evolution](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/75/02-alignment-pdf-48-2048.jpg)

![Heuristic Methods
• the algorithms we learned today take O(nm) time to align
sequences, which is too slow for searching large databases
– imagine an internet search engine, but where queries and results
are protein sequences
• heuristic methods do fast approximation to dynamic
programming
– example: BLAST [Altschul et al., 1990; Altschul et al., 1997]
– break sequence into small (e.g. 3 base pair) “words”
– scan database for word matches
– extend all matches to seek high-scoring alignments
– tradeoff: sensitivity for speed](https://image.slidesharecdn.com/02-alignment-231003125704-9f36112d/75/02-alignment-pdf-50-2048.jpg)

















































