Dynamic programming and pairwise sequence alignment
This document discusses pairwise sequence alignment algorithms. It describes the Needleman-Wunsch and Smith-Waterman algorithms for global and local sequence alignment, respectively. These algorithms construct scoring matrices to find the optimal alignment between two sequences by maximizing matches and minimizing gaps. BLAST is also summarized as a tool for comparing nucleotide and protein sequences through database searches. Applications of pairwise sequence alignment include gene characterization and determining molecular evolutionary distances.
Introduction
Algorithm used indynamic programing
• Needleman-Wunsch algorithm
• Smith- Waterman algorithm
Pairwise sequence alignment
• BLASTn
• BLASTp
Applications of pairwise sequence alignment
References
1 November 2019 2
3.
Dynamic programmingis a computational method that is
used to align two proteins or nucleic acids sequences.
This method is very important for sequence analysis because
it provides the very best or optimal alignment between
sequences.
Alignment includes matched, mismatched characters and
gaps in the two sequences that positioned so that the
number of matches between identical or related character is
maximum as possible.
INTRODUCTION
1 November 2019 3
4.
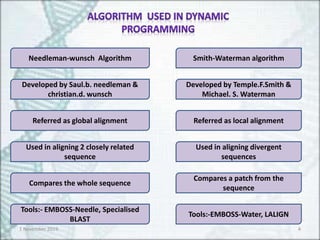
Needleman-wunsch Algorithm
Developed bySaul.b. needleman &
christian.d. wunsch
Referred as global alignment
Used in aligning 2 closely related
sequence
Compares the whole sequence
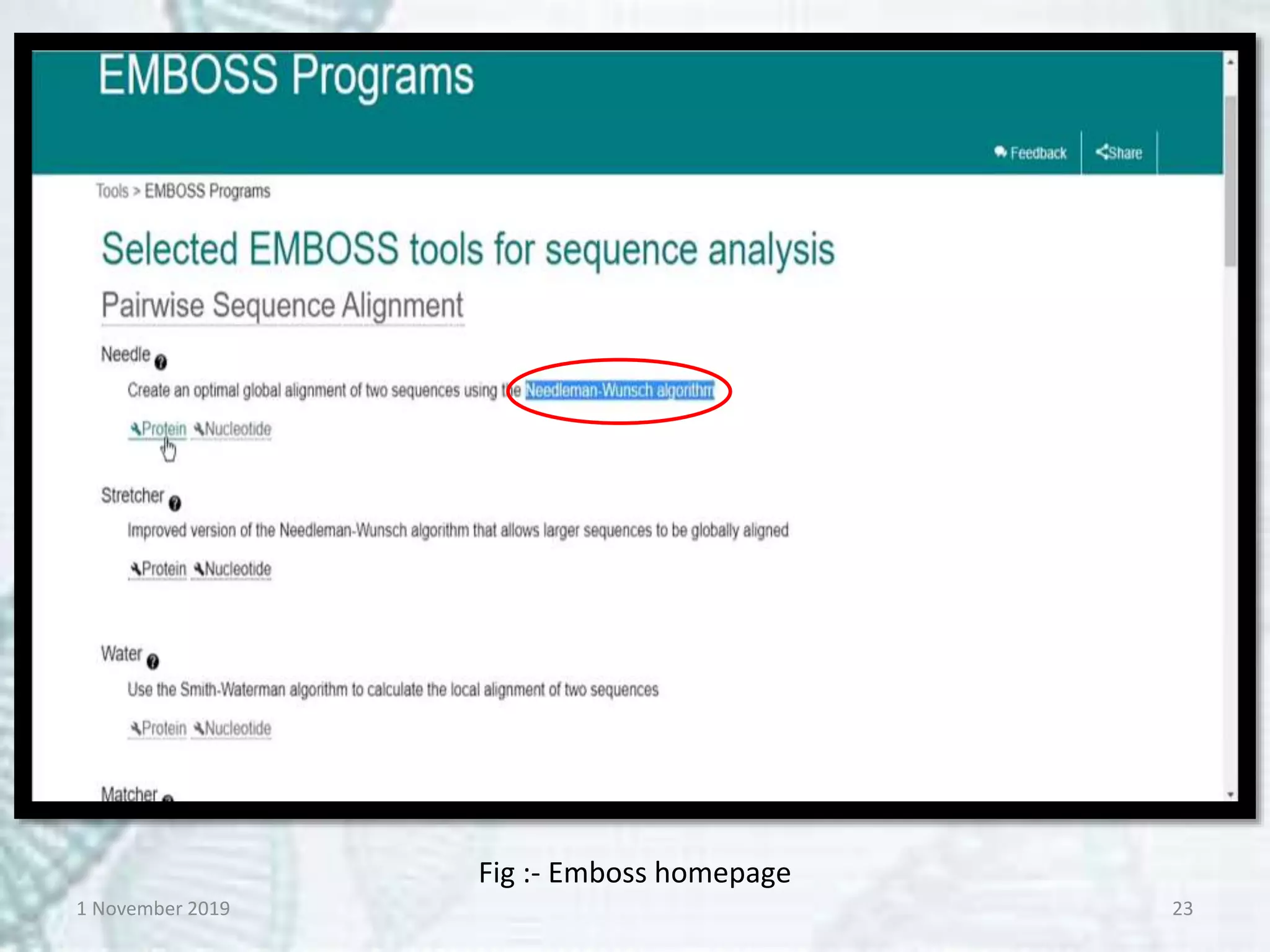
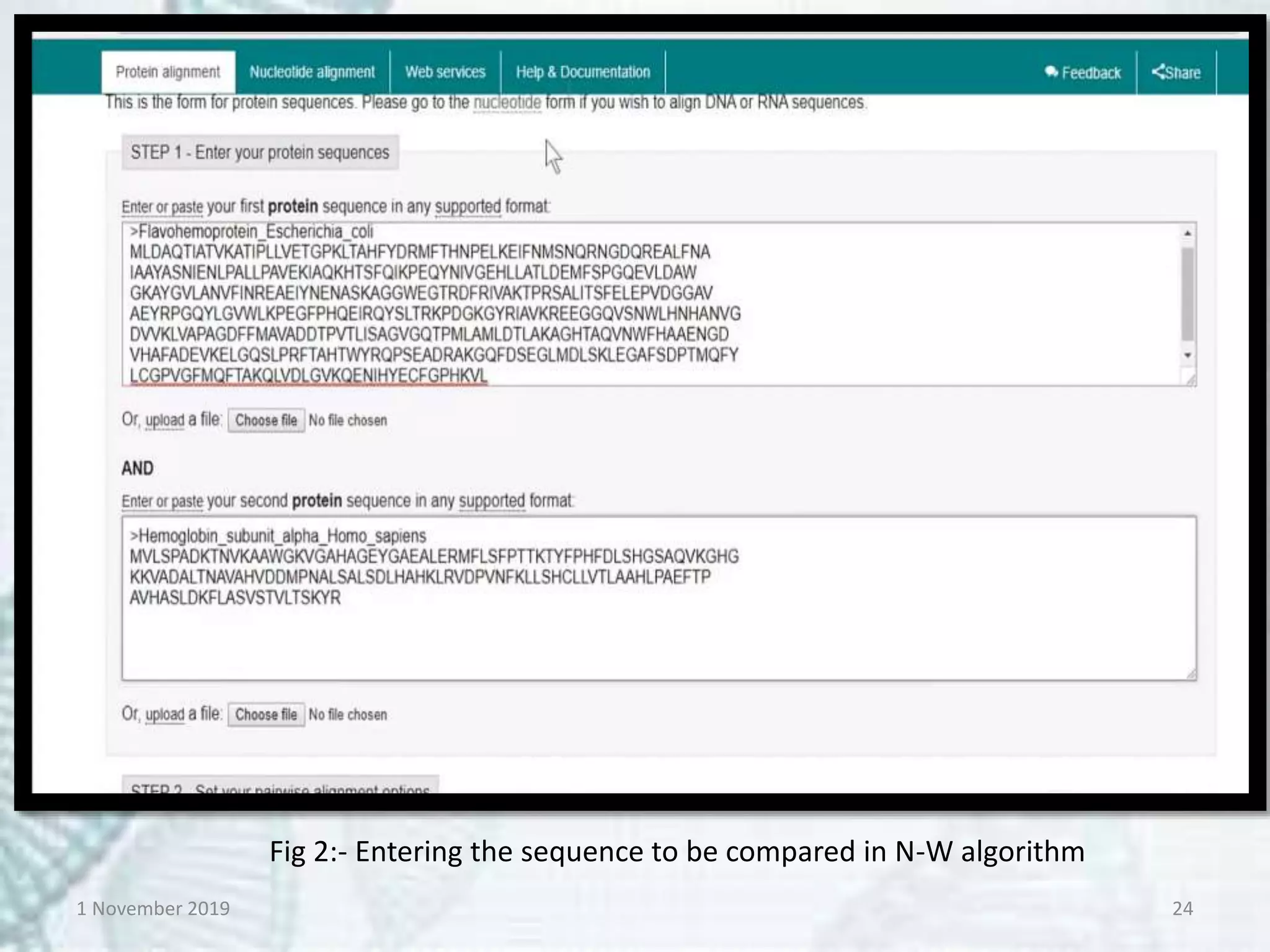
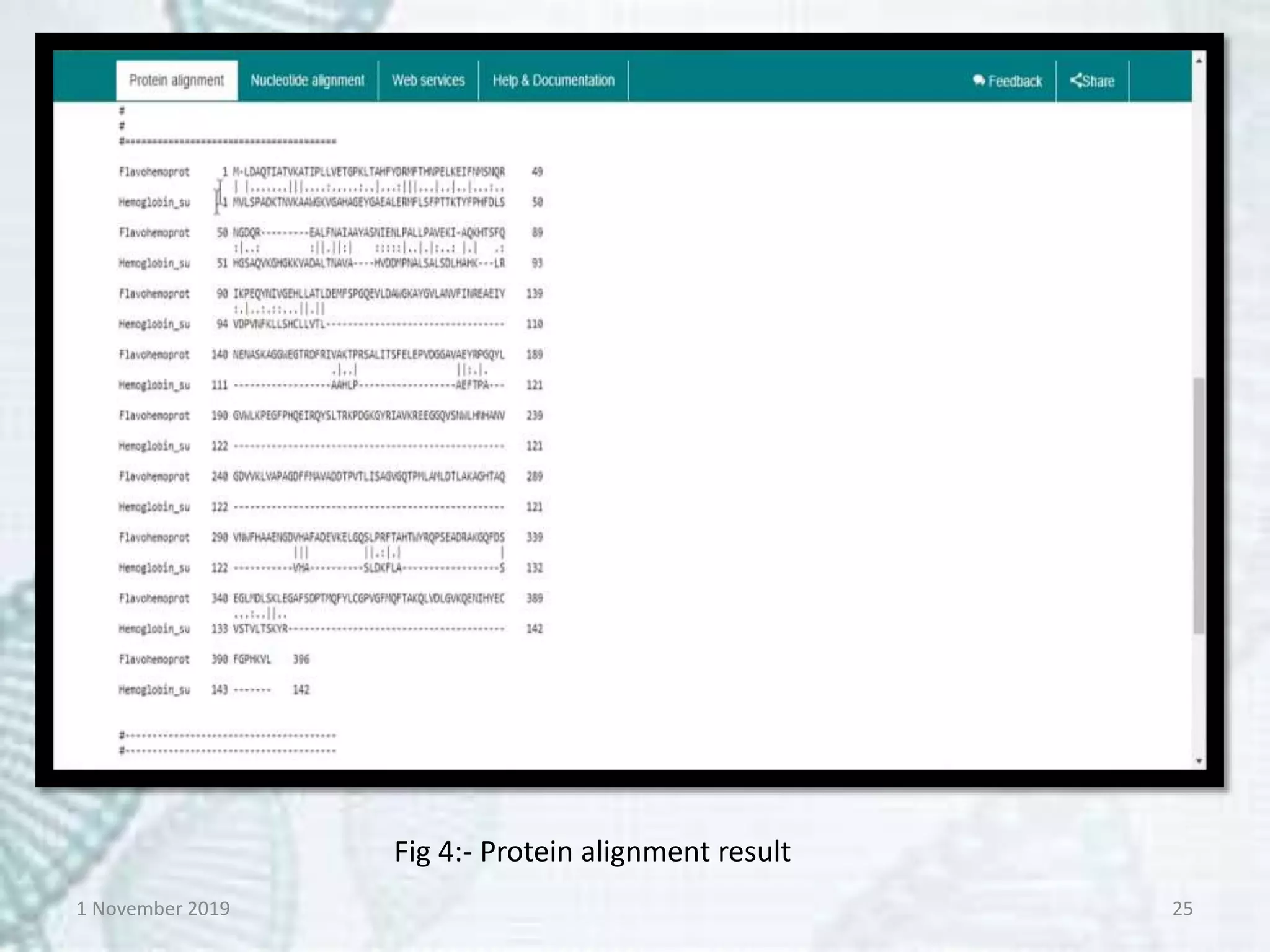
Tools:- EMBOSS-Needle, Specialised
BLAST
Smith-Waterman algorithm
Developed by Temple.F.Smith &
Michael. S. Waterman
Referred as local alignment
Used in aligning divergent
sequences
Compares a patch from the
sequence
Tools:-EMBOSS-Water, LALIGN
1 November 2019 4
5.
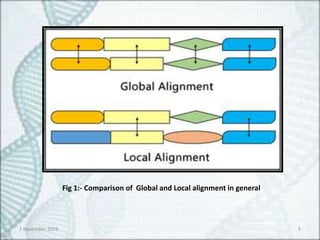
Fig 1:- Comparisonof Global and Local alignment in general
1 November 2019 5
6.
Needleman-Wunsch algorithm
• Alignsprotein or nucleic acid sequences
• Divides a large problem into a series of smaller
problems
• Uses the solution of smaller problem to
reconstruct a solution to the larger problems
1 November 2019 6
7.
Constructing the matrix
Wewill have 2 matrices of 2D representation viz,
1. The score matrix
2. Traceback matrix
The N-W algorithm consists of 4 steps:-
1. Initialization of the score matrix
2. Filling up the matrix
3. Traceback
4. Alignment
1 November 2019 7
8.
1.Initializing the scoringmatrix
G C A T G C
0 -1 -2 -3 -4 -5 -6
G -1
T -2
A -3
C -4
G -5
C -6
Match = 1
Mismatch. = -1
Gap. = -1
1 November 2019 8
9.
2. Filling thematrix
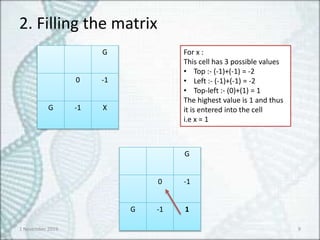
G
0 -1
G -1 X
For x :
This cell has 3 possible values
• Top :- (-1)+(-1) = -2
• Left :- (-1)+(-1) = -2
• Top-left :- (0)+(1) = 1
The highest value is 1 and thus
it is entered into the cell
i.e x = 1
G
0 -1
G -1 1
1 November 2019 9
10.
Contd..
G C
0 -1-2
G -1 1 X
C -2 Y
For X :
Top: (-2)+(-1) = (-3)
Left: (+1)+(-1) = (0)
Top-Left: (-1)+(-1) = (-2)
For Y :
Top: (1)+(-1) = (0)
Left: (-2)+(-1) = (-3)
Top-Left: (-1)+(-1) = (-2)
The highest value for X and Y is 0,
thus it is entered into the cell.
i.e X = O; Y = 0
G C
0 -1 -2
G -1 1 0
C -2 0
1 November 2019 10
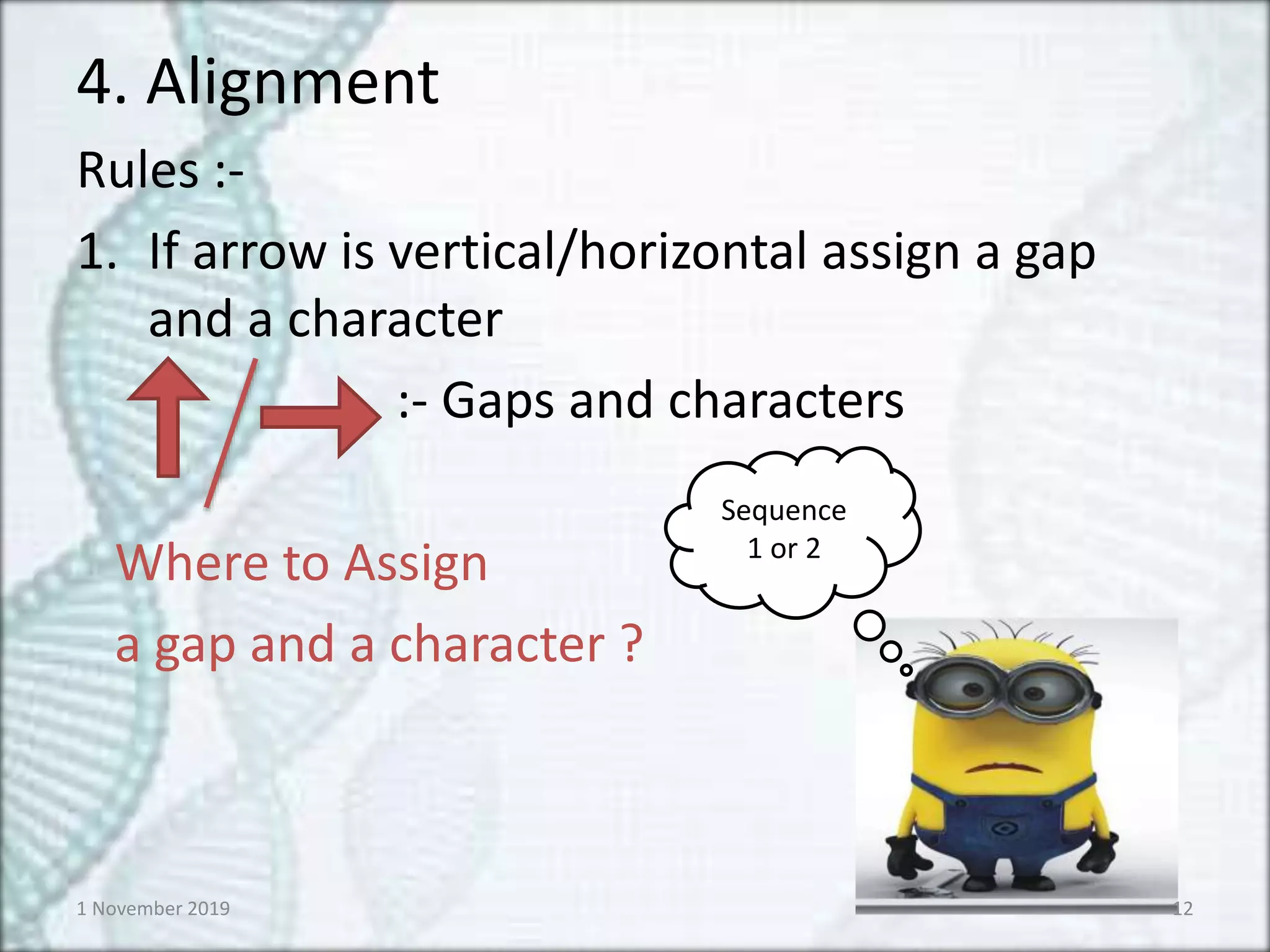
4. Alignment
Rules :-
1.If arrow is vertical/horizontal assign a gap
and a character
:- Gaps and characters
Where to Assign
a gap and a character ?
Sequence
1 or 2
1 November 2019 12
13.
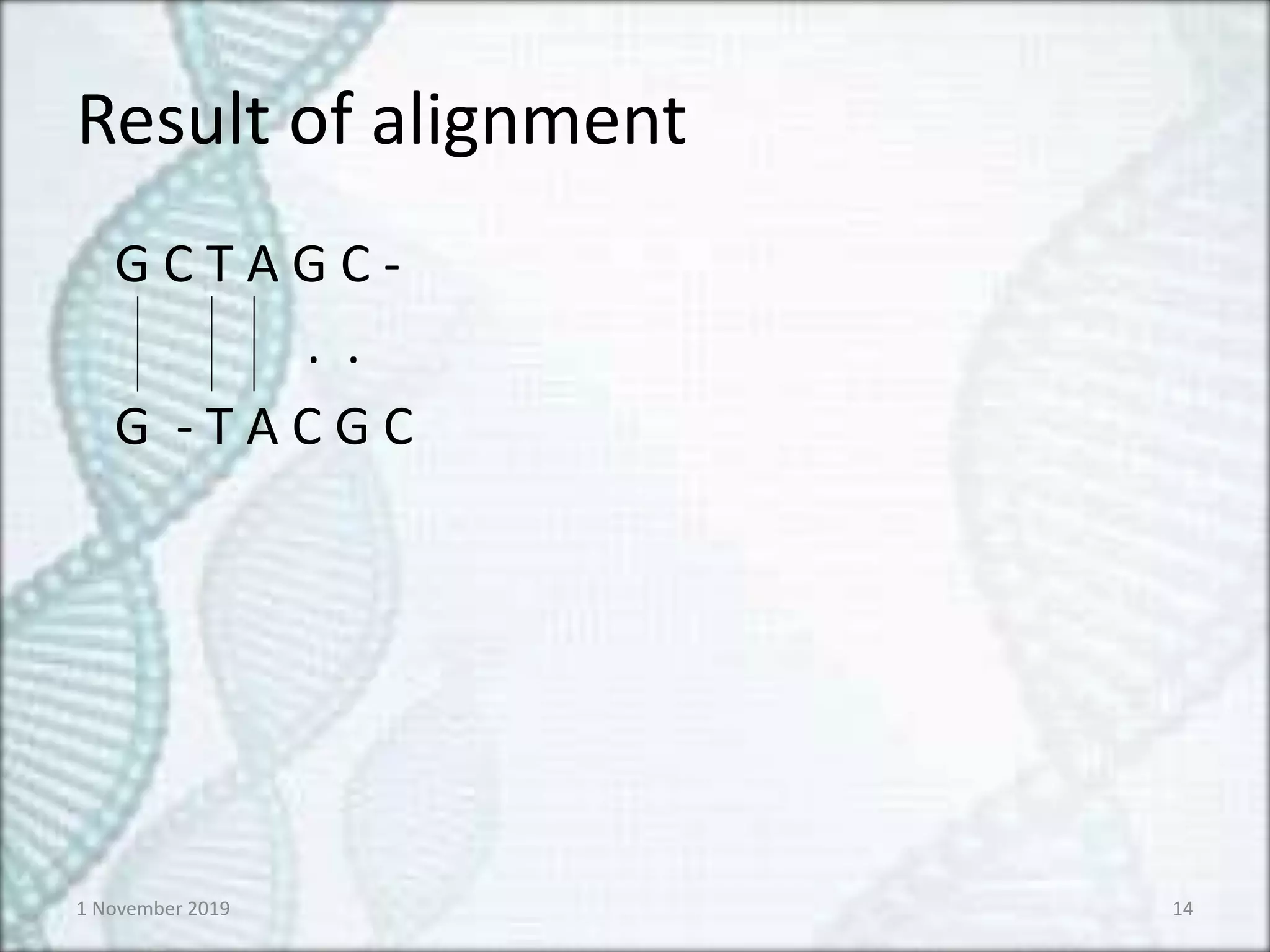
Contd…
Ans) The gapwill be assigned in the direction of
the arrow and the character will be assigned in
the opposite direction
2. If there is a diagonal arrow both the
characters will be assigned
:- Both characters
1 November 2019 13
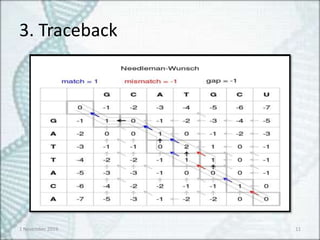
• S-W algorithmis modified version of
Needleman- Wunsch algorithm
• Negative scoring matrix cells are set to zero
• Traceback procedure starts at the highest
scoring matrix cells and procedure until a cell
with score zero is found
1 November 2019 15
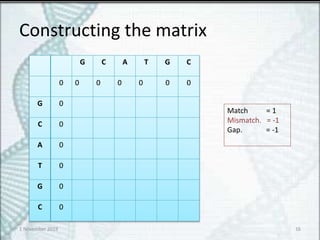
16.
Constructing the matrix
GC A T G C
0 0 0 0 0 0 0
G 0
C 0
A 0
T 0
G 0
C 0
Match = 1
Mismatch. = -1
Gap. = -1
1 November 2019 16
17.
Contd..
G C
0 -1-2
G -1 1 X
C -2 Y
For X :
Top: (-2)+(-1) = (-3)
Left: (+1)+(-1) = (0)
Top-Left: (-1)+(-1) = (-2)
For Y :
Top: (1)+(-1) = (0)
Left: (-2)+(-1) = (-3)
Top-Left: (-1)+(-1) = (-2)
The highest value for X and Y is 0,
thus it is entered into the cell.
i.e X = O; Y = 0
G C
0 -1 -2
G -1 1 0
C -2 0
1 November 2019 17
When comparing 2sequences it is Pairwise
sequence alignment (nucleic acids or protein)
When comparing more than 2 sequence it is
Mulitiple sequence alignment (nucleic acids
or protein)
1 November 2019 26
27.
Contd…
Pairwise sequencealignment is concerned
with comparing 2 DNA or 2 Amino acids
sequences
For ex.
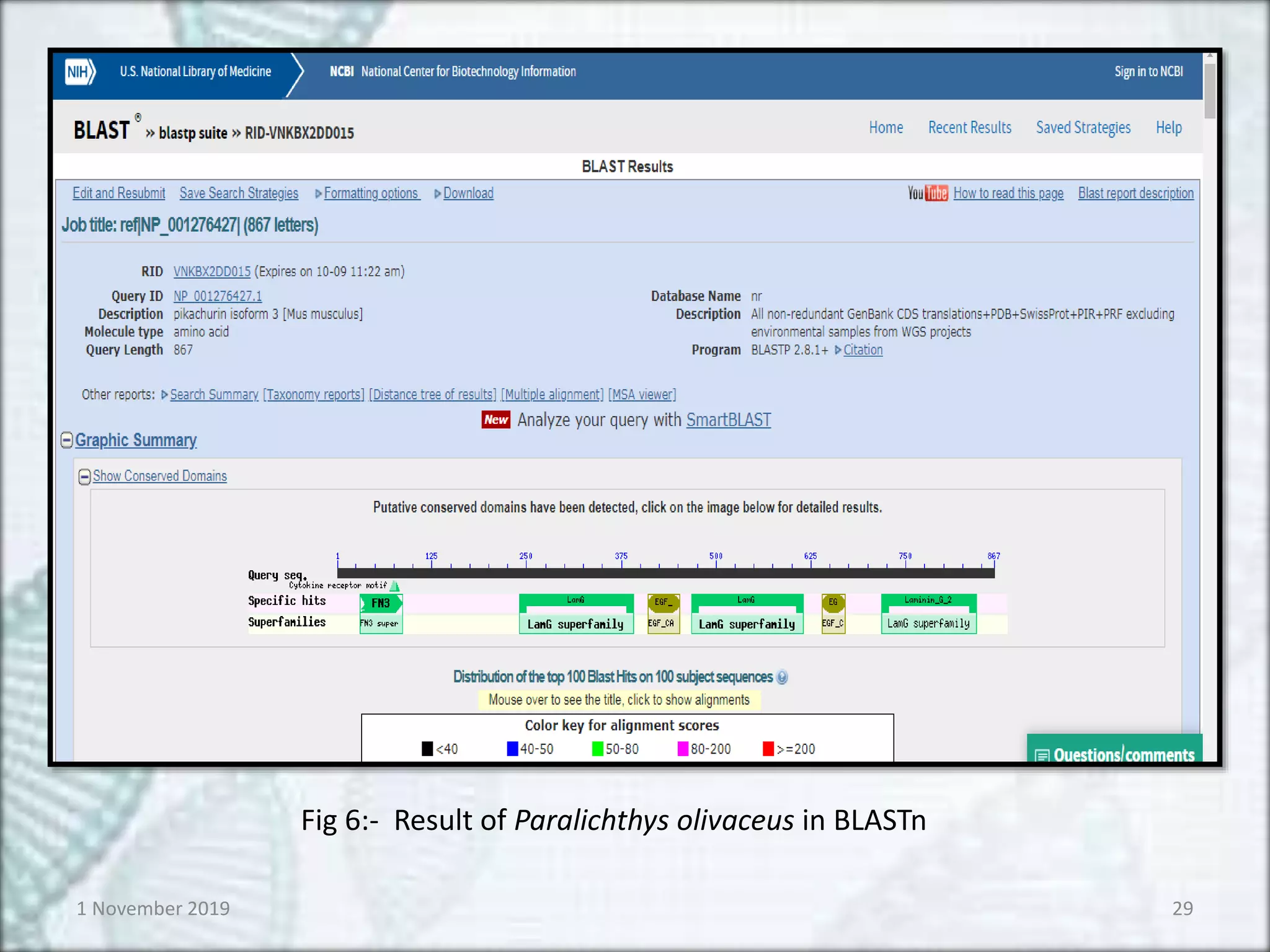
BLASTn :- for nucleotide sequence
BLASTp :- for protein sequence
1 November 2019 27
28.
BLAST isa Basic Local Alignment Search Tool.
Used for comparing primary biological
information viz,
–Amino acid sequences of protein
–Nucleotide of DNA or RNA sequences
BLASTn and BLASTp is particularly used for
comparing nucleotide sequence and protein
sequence respectively1 November 2019 28
29.
1 November 201929
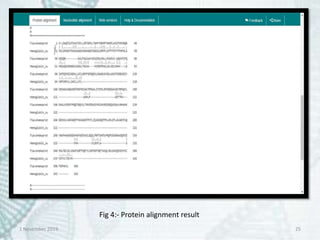
Fig 6:- Result of Paralichthys olivaceus in BLASTn
30.
1 November 201930
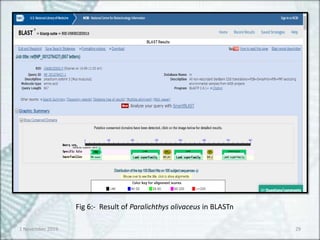
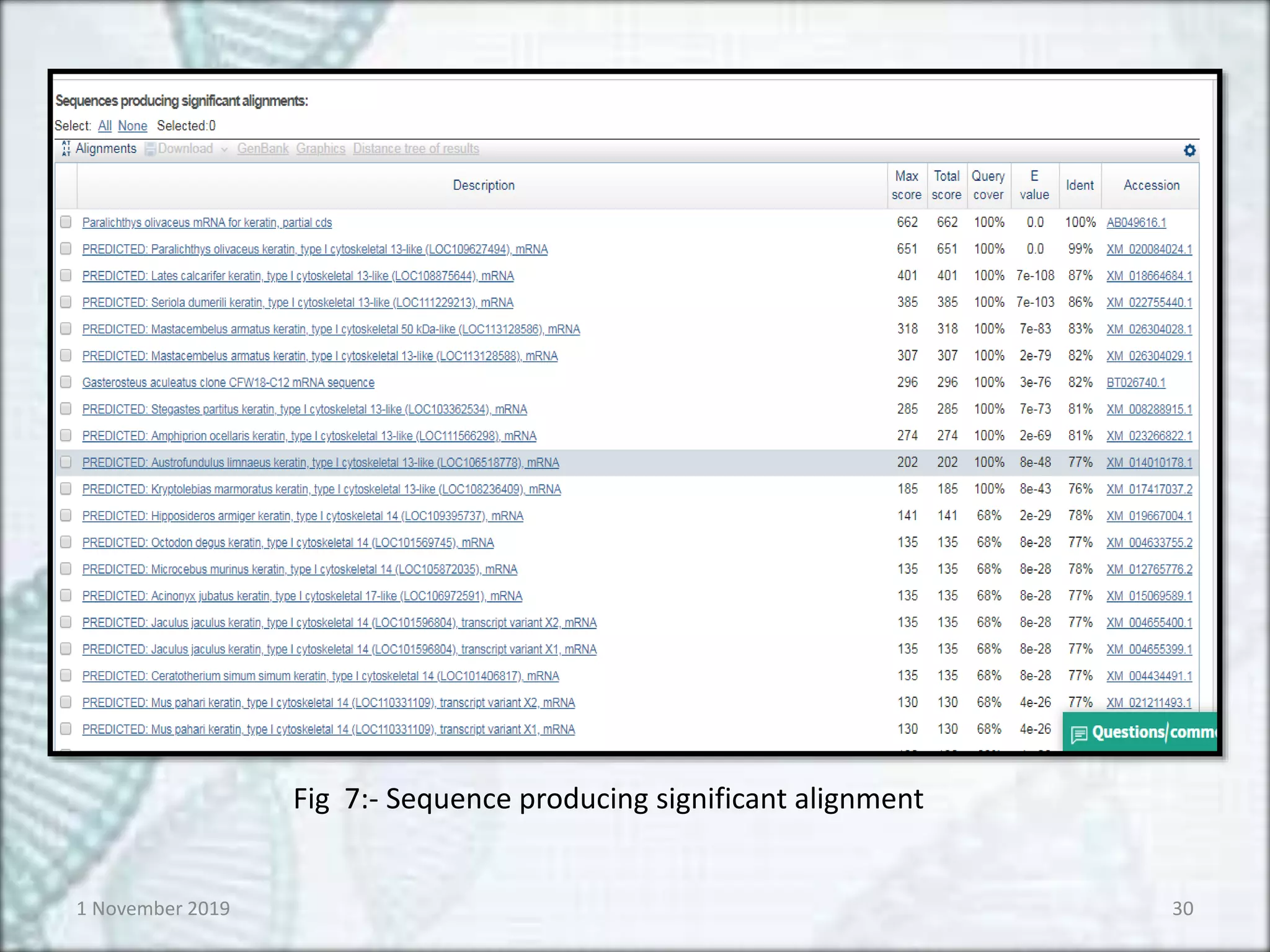
Fig 7:- Sequence producing significant alignment
31.
1 November 201931
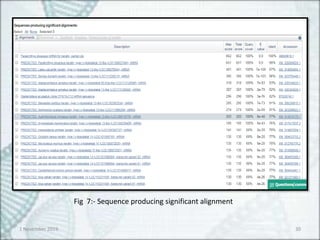
Fig 8:- Sequence alignment of Paralichthys olivaceus
32.
Searching largesequences for matches
Characterize newly sequenced genes or gene
products
Molecular distance of evolution between
species
1 November 2019 32
33.
REFERENCE
Introduction ofNeedleman-wunsch and smith
waterman algorithm
David Mount;“Bioinformatics sequence and
genome analysis”,chp 3;pp53
Needleman,S.B.and wunsch,C.D.(1970), “A
general method applicable to the search for
similarities in the amino acid sequence of two
proteins”, J.Mol.Biol.,vol,48,pp 443-453.
1 November 2019 33
34.
Contd..
Smith, T.t.&Waterman,M.S.(1981), “Identification of
common molecular subsequences”, -
J.Mol.Biol.,vol.147,pp195-197
Tools used for sequence alignment
(https://www.omictools.com)
(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3446
765/)
1 November 2019 34