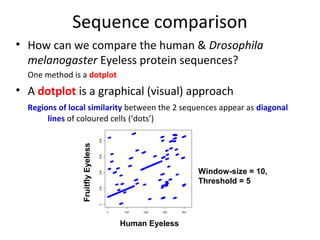
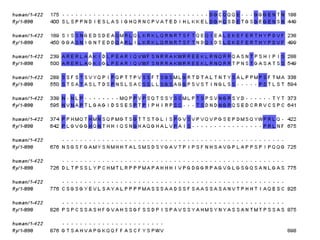
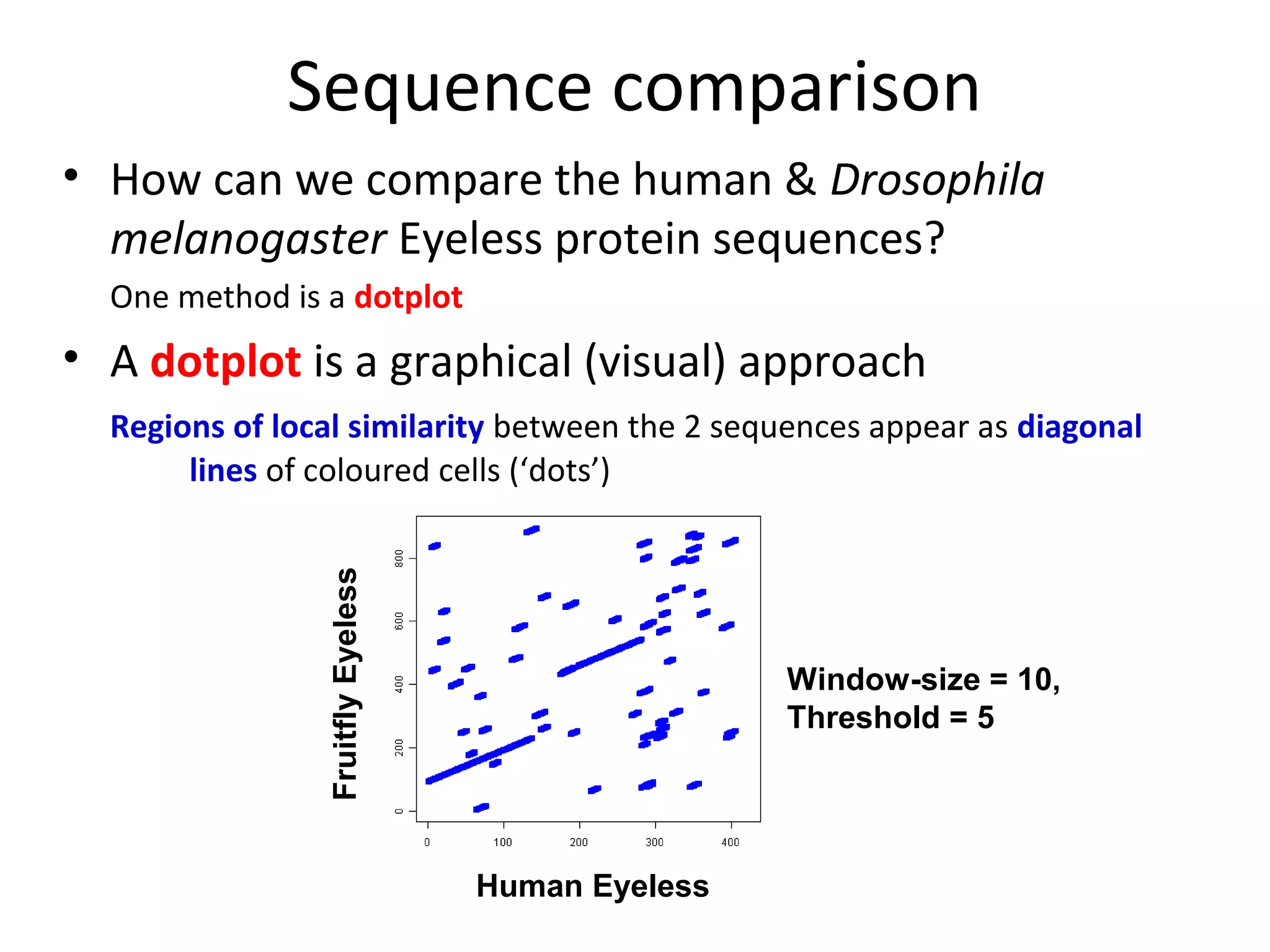
1) Pairwise sequence alignment is a method to compare two biological sequences like DNA, RNA, or proteins. It involves arranging the sequences in columns to highlight their similarities and differences.
2) There are many possible alignments between two sequences, but most imply too many mutations. The best alignment minimizes the number of mutations needed to explain the differences between the sequences.
3) For short protein sequences like "QKGSYPVRSTC" and "QKGSGPVRSTC", the optimal alignment implies one single mutation occurred since the sequences diverged from a common ancestor.